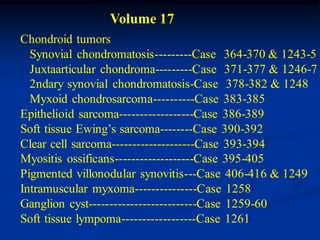
Volume 17
•
5 likes•1,144 views

The document describes several cases of rare soft tissue tumors, including: 1) Four cases of epithelioid sarcoma affecting the foot, forearm, and wrist in patients aged 22-36, which present as ulcerated lesions and are difficult to diagnose. 2) Three cases of myxoid chondrosarcoma in the thigh, shoulder, and leg in patients aged 41-65, which appear similar to hematomas and have a characteristic myxoid and chordoid pathology. 3) Several cases of synovial chondromatosis, juxta-articular chondroma, and secondary synovial chondromatosis affecting various large joints in patients aged 13

Recommended
More Related Content
What's hot
What's hot (20)
Viewers also liked
Similar to Volume 17
Recently uploaded
Recently uploaded (20)
Volume 17
- 1. Volume 17 Chondroid tumors Synovial chondromatosis---------Case 364-370 & 1243-5 Juxtaarticular chondroma---------Case 371-377 & 1246-7 2ndary synovial chondromatosis-Case 378-382 & 1248 Myxoid chondrosarcoma----------Case 383-385 Epithelioid sarcoma------------------Case 386-389 Soft tissue Ewing’s sarcoma--------Case 390-392 Clear cell sarcoma--------------------Case 393-394 Myositis ossificans-------------------Case 395-405 Pigmented villonodular synovitis---Case 406-416 & 1249 Intramuscular myxoma---------------Case 1258 Ganglion cyst--------------------------Case 1259-60 Soft tissue lympoma------------------Case 1261
- 4. Synovial Chondromatosis Synovial chondromatosis is a rare dysplasia seen in younger patients. It is associated with metaplastic cartilage within the synovial lining of major joints such as the hip, knee, shoulder and elbow. It is twice as common in males as females and usually occurs in patients in the 20-40 age group. It is a monarticular disease that presents with symptoms of crepitation in the affected joint with mild, intermittent effusion sometimes associated with pain. Because of chronic irritation to the joint and damage to the articular cartilage, osteoarthritis is an ultimate problem with this disease and can lead to a total joint replacement at a later age. In rare instances this condition can mutate into a secondary chondro- sarcoma, usually around the hip or knee joint but seldom in the shoulder area. This usually occurs in the later years of life. In the early stages before the cartilage becomes calcified, the synovial chondromatosis may be difficult to pick up on routine radiographic examination. As time passes, the cartilage begins
- 5. to calcify in a typical chondroid pattern that suggests the diagnosis of a chondroid tumor in or about a major joint. With excessive proliferation, the cartilage can extrude out of the joint into adjacent soft tissue, similar to what occurs with pigmented villonodular synovitis. As the disease progresses, it is not unusual to see enchondral ossification occurring within the cartilage when it is still attached to the synovial lining and has access to a blood supply. Multiple loose bodies are common with this disease and can run as high as 200 pellets within a major joint that sometimes aggregrate into a large mass that has the appearance of a chondrosarcoma. Treatment consists of a surgical resection of the loose bodies as well as a subtotal synovectomy of the tissue that produces the loose bodies. Multiple surgical procedures may be required because of a high recurrence rate. As in PVNS, it is not unusual to see a solitary focus of synovial chondromatosis with the remaining synovial lining being normal in appearance with only a solitary mass of cartilage attached to the synovial lining. This localized nodular form
- 6. is more common about the hip, knee and ankle area. It is very common to find loose bodies in a major arthritic joint in older patients secondary to osteoarthritis where the joint cartilage is broken away from the joint surface and than becomes reattached to the synovial lining and gives the pseudo-appearance of primary synovial chondromatosis when, in fact, the primary etiology in this so-called secondary form is degenerative osteoarthritis. The secondary form is seen in patients past the age of 50 years, where- as the primary dysplastic form arising from the synovial lining is seen during the first three to four decades of life.
- 7. CLASSIC Case #364 33 year male with synovial chondromatoasis of shoulder
- 8. Sagittal T-2 MRI showing multiple calcified cartilage bodies
- 9. Coronal T-2 MRI showing loose bodies
- 10. Surgical exposure showing loose bodies
- 11. Loose bodies and synovial lining following surgery
- 12. Cartilaginous bodies arising from synovial lining
- 13. Cartilaginous body formation in synovial villus
- 14. Case #365 26 year male with synovial chondromatosis knee joint
- 15. AP x-ray
- 16. Gross appearance of synovial chondromatosis
- 17. Microscopic evidence of cartilage elements in synovium
- 18. Case #366 28 year female with synovial chondromatosis knee
- 19. Another view
- 20. Photomic showing cartilage in synovial lining
- 21. Case #366.1 63 year female with long history of synovial chondromatosis
- 22. Sag T-2 Sag Gad
- 23. Cor T-2 Axial Gad
- 24. Case #366.2 Synovial chondromatosis 42 year female with grinding sensations in knee for years
- 25. Case #367 38 year male with synovial chondromatosis left hip
- 26. Frog leg lateral
- 27. Coronal T-2 MRI
- 28. Sagittal T-2 MRI cut showing loose bodies
- 29. Surgical removal of cartilage bodies
- 30. Photo of largest cartilage body
- 31. Case #368 24 year female with large cartilaginous pelvic mass arising from synovial chondromatosis of left hip
- 32. tumor Close up of deformed femoral neck
- 33. tumor Note deformity of medial femoral neck
- 34. femoral N Exposure of large pelvic tumor by removal ant pubic ramus
- 35. pubic groove Resected cartilage masses and loose hip joint bodies
- 36. Cut specimen of one mass formed from compressed pellets
- 37. Loose bodies from hip joint
- 38. cartilage Photomic of cartilage forming in synovial lining
- 39. Post op x-ray showing ramus surgical defect
- 40. 9 years later with DOA requiring THA
- 41. Case #369 59 year male with synovial chondromatosis right hip for 20 years
- 42. 5 years later with 2ndary chondrosarcoma in femoral neck
- 43. Frog leg lateral showing chondrosarcoma
- 44. Femoral head & neck specimen from THA showing chondrosarcoma in lower neck area
- 45. chondrosarcoma Synovectomy specimen obtained at time if THA
- 47. 1 yr later we see recurrent tumor requiring hemipelvectomy
- 48. Case #370 50 year female with 25 year history of synovial chondromatosis
- 49. Coronal T-2 MRI several years later with 2ndary chondrosarc
- 50. Dedifferentiated Case #370.1 Cor T-1 T-2 chondrosarcoma 60 yr male with swollen knee for years with recent increase in size
- 51. Cor T-2 Gad
- 52. Sag T-2 Gad T-2
- 53. Axial T-2 Gad
- 54. Case #1243 20 year female with synovial chondromatosis hip
- 55. Frog leg lateral
- 56. CT scan showing loose bodies
- 57. Case #1244 26 year female with synovial chondromatosis hip
- 58. CT scan
- 59. CT scan at lower level
- 60. Photomic showing embryonic cartilage in synovial lining
- 61. Case #1245 49 year female with synovial chondromatosis wrist
- 62. AP view
- 63. Axial Gad contrast MRI
- 64. loose pellets fluid Sagittal T-1 MRI showing wrist effusion and loose pellets
- 66. CLASSIC Case #371 25 year old with juxta-articular chondroma posterior knee
- 67. AP view
- 71. Photomic
- 72. Case #372 41 year male with juxta-articular chondroma ant knee
- 73. Sagittal T-1 MRI
- 74. Sagittal T-2 MRI
- 75. Axial T-2 MRI
- 76. Photomic
- 77. Case #372.1 Juxtr-articular chondroma 60 yr female with lump in anterior knee area for years
- 78. Sag T-1 T-2 Gad
- 79. Axial T-1 PD Gad
- 80. Case #373 58 year male with juxta-articular chondroma posterior knee
- 81. Sagittal T-2 MRI
- 82. Axial T-2 MRI
- 83. Case #374 Sagittal T-1 MRI tumor 37 year male with juxta-articular chondroma post knee
- 84. Sagittal T-2 MRI
- 85. tumor Sagittal gad contrast MRI with rim enhancement
- 87. tumor Axial gad contrast with rim enhancement
- 88. Case #1248 36 year male with soft tissue chondrosarc behind knee
- 89. Sagittal T-2 MRI
- 90. Axial T-1 MRI
- 91. Axial T-2 MRI
- 92. Case #375 13 year male with juxta-articular chondroma posterior knee
- 93. Case #376 18 year male with juxta-articular chondroma ankle
- 94. Case #376.1 Juxta-articular chondroma 38 old male with long history of catching sensations in ankle
- 95. Axial T-1 PD Gad
- 96. Sag T-1 PD Gad
- 97. Case #377 19 year male with tenosynovial chondromatosis
- 98. Lateral view
- 99. Photomic
- 100. Case #1247 44 year male with solitary synovial chondroma hip
- 101. Lateral view
- 102. Photomic from resected specimen
- 103. Secondary Synovial Chondromatosis
- 104. CLASSIC Case #378 49 year female with 2ndary synovial chondromatosis from osteoarthritis of knee AP x-ray
- 105. Lateral view
- 106. Sagittal T-2 MRI
- 107. Axial proton density MRI fluid
- 108. Axial T-2 MRI
- 109. Case 379 Coronal T-1 MRI osteoarthritis 76 year male with 2ndary synovial chondromatosis knee
- 110. Sagittal T-2 MRI showing loose bodies in popliteal synovial cyst
- 111. Axial T-2 MRI with loose body in popliteal cyst
- 112. Axial T-2 MRI with many loose bodies in popliteal cyst
- 113. Case #379.1 Secondary synovial chondromatosis 57 year old soccer player with prior history of knee surgery
- 114. Sag T-1 T-2 Gad
- 115. Axial T-2 Gad
- 116. Case #380 70 year female 2ndary synovial chondromatosis from osteoarthritis knee
- 117. Lateral view
- 118. Case #381 55 year female 2ndary synovial chondromatosis from osteoarthritis knee
- 119. Lateral view
- 120. Sagittal proton density MRI showing loose bodies in popliteal cyst
- 121. Case #381.1 Secondary synovial osteochondromatosis 62 year old male with DOA and large necrotizing mass over tibia
- 122. Sag T-1 T-2 Gad
- 123. T-2 Gad Axial T-2 Gad
- 124. Post op surgical debriedment Post op flap coverage over tibia
- 125. Case #382 69 year male with 2ndary synovial chondromatosis from traumatic arthritis of elbow
- 126. Surgical removal of loose bodies
- 127. Loose bodies and pieces of synovium
- 128. Case #1246 46 year female 2ndary synovial chondromatosis talonarvicular second to old trauma of subtalar joint
- 129. Sagittal T-1 MRI showing traumatic arthritic changes
- 130. Sagittal T-2 MRI showing excessive synovial fluid
- 131. Coronal T-2 MRI
- 133. Myxoid Chondrosarcoma (Chordoid Sarcoma) The extra-skeletal myxoid chondrosarcoma is a very rare soft tissue tumor in the deep muscle belly, occurring most often in the extremities in patients over 40 years of age. Males are affected twice as often as females. The tumor is slow growing and may cause local pain and tenderness. Common locations are the thigh, popliteal fossa, and shoulder girdle. It presents with the clinical appearance of a myxoid liposarcoma. Pathologically, the tumors are greyish to tannish brown, depending on the amount of hemorrhage into the tumor. Because hemorrhage often occurs, it can be mistaken for a hematoma. Histologically, the tumor has a myxoid appearance with chords and nests of anastomosing cells that have a chondroblastic appearance. The histology is very similar to that of chordoma of the sacrum. The tumor is considered low grade in most instances; it is slow growing but has the potential
- 134. for local recurrence and pulmonary metastases in about one-third of cases. Treatment consists of aggressive wide local resection or amputation, if needed, followed by local radiation therapy. Chemo- therapy is usually not indicated.
- 135. CLASSIC Case #383 Coronal T-1 MRI tumor 55 year male with myxoid chondrosarcoma distal thigh
- 136. Coronal T-2 MRI tumor
- 137. tumor Axial T-2 MRI
- 138. Resected hemorrhagic surgical specimen
- 139. Photomic showing chordoid myxoid pattern like chordoma
- 140. Pathology similar to that of a chordoma
- 141. Case #384 Sagittal T-2 MRI tumor 41 year female with myxoid chondrosarcoma shoulder
- 142. tumor Axial gad contrast MRI
- 143. Case #385 65 year male with soft tissue myxoid chondrosarcoma leg eroding tibia AP view
- 144. Lateral view
- 145. Sagittal T-2 MRI tumor
- 146. tumor Axial T-2 MRI
- 147. tumor Another axial T-2 MRI
- 148. Epithelioid Sarcoma
- 149. Epithelioid Sarcoma The epithelioid sarcoma affects young adults and is most commonly seen in the fingers, hand and forearm where it is considered the most common soft tissue sarcoma next to the alveolar rhabdomysarcoma and synovial sarcoma. It can also occur in the popliteal area, the buttock, thigh, shoulder, foot and ankle area. It affects twice as many males as females. These tumors are frequently misdiagnosed as a benign granulomatous process and are often attached to tendon sheaths and facial planes with associated cutaneous ulcerations that may be multiple in nature. Calcification or even bone formation can occur in about 15% of cases. The histological appearance of this lesion displays a distinct nodular growth pattern with epithelioid nests of cells at the center surrounded by lymphocytic infiltration. The differential diagnosis would include necrotizing infectious diseases such as tuberculosis or granuloma annulare or rheumatoid nodules. Regional lymph
- 150. node involvement occurs in about 35% of cases and metastases to the lung in about 50% of cases. Because of the benign clinical appearance of this lesion, it is common for surgeons to attempt local resection but there is a high recurrence rate that eventually leads to amputation. Local radiation therapy can help to decrease the chance of local recurrence.
- 151. CLASSIC Case #386 36 year male with epithelioid sarcoma foot ulceration
- 152. Side view with ulcerated cap
- 153. tumor CT scan
- 154. tumor Another CT cut
- 155. necrotic center Scanning lens photomic
- 156. Close up outer edge
- 157. Higher power of epithelioid cells
- 158. Case #387 34 year male with epithelioid sarcoma forearm Sagittal T-2 MRI
- 159. Sagittal Gad contrast MRI tumor
- 160. tumor Axial T-2 MRI
- 161. necrosis tumor Axial gad contrast MRI
- 162. Photomic showing epithelioid cells
- 163. Case #387.1 Epithelioid sarcoma 22 year old male with spontaneous fungating wound volar wrist
- 164. Cor T-1 T-2 FS Gad
- 165. Sag T-1 T-2 FS Gad
- 166. Axial T-1 T-2 FS Gad
- 167. Case #388 27 year female with epithelioid sarcoma sole of foot
- 168. Case #388 Sagittal T-1 MRI
- 169. Axial proton density MRI
- 170. Axial T-2 MRI tumor
- 171. Case #389 50 year male with epithelioid sarcoma middle finger
- 173. Extra-skeletal Ewing’s Sarcoma Ewing’s sarcoma is usually associated with primary tumor of bone but in a small percentage of cases, Ewing’s sarcoma can occur in soft tissue completely unattached to the skeletal system. However, the histological appearance and the clinical picture associated with soft tissue Ewing’s sarcoma is basically the same as that of skeletal Ewing’s. This condition is seen in patients between the age of 15 and 30 years. It occurs in males and females equally and is rare in black patients. The most common location is the chest wall, followed by the lower extremities, paraspinous area, pelvis, hip and retroperitoneum. The least common location is the upper extremity. The reciprocal translocation of the long arm of chromosomes 11 and 22 is seen in soft tissue Ewing’s, just as it is in skeletal Ewing’s. The prognosis for five year survival is approximately 65%, similar to that of skeletal Ewing’s. Treatment consists of wide local resection when possible, followed by
- 174. local radiation therapy if indicated. Adjuvant chemotherapy is commonly used because of the excellent response, similar to that of skeletal Ewing’s sarcoma.
- 175. CLASSIC Case #390 28 year female with soft tissue Ewing’s sarcoma anterior thigh Coronal proton density MRI
- 177. Coronal T-2 MRI tumor
- 178. tumor Axial T-2 MRI
- 179. Surgical specimen inked and cut in path lab
- 180. Photomic
- 181. Case #390.1 Axial T-1 MRI 38 year female with painful thigh mass 2 months
- 182. Axial T-2 Axial Gad necrosis
- 183. necrosis Coronal T-2 Coronal Gad contrast
- 184. Case #391 14 year male with soft tissue Ewing’s sarcoma distal thigh
- 185. Axial proton density MRI tumor
- 186. Axial T-2 MRI tumor
- 187. Photomic
- 188. Case #391.1 T-1 Sag MRI T-2 Sag 18 yr female with Ewing’s sarcoma in sciatic nerve looking like a benign neurilemoma
- 189. Axial Gad MRI at two different levels Lower level Upper level
- 190. Coronal Gad MRI
- 191. Case #392 16 year female with soft tissue Ewing’s sarcoma thigh
- 192. Coronal T-1 MRI
- 193. tumor Axial T-2 MRI
- 194. Clear Cell Sarcoma
- 195. Clear Cell Sarcoma The clear cell sarcoma is thought to be a deep, non-cutaneous variant of the pigmented melanoma. It is a very rare tumor affecting females more than males. It is typically seen between the ages of 20 and 40 years. It usually occurs in tendon sheaths and fascial planes, especially around the foot and ankle area, similar to the clinical appearance seen with synovial sarcoma with which it can be confused. The tumor usually begins as a slow growing lump that has a benign appearance but after a period of several years the tumor will start growing more rapidly and become painful. It has a high potential to metastasize to local lymph nodes and to the lung. Approximately 50% of patients with this tumor will be dead in five years. The microscopic appearance is similar to that of the epithelioid sarcoma, especially if melanin is not found in the specimen. Treatment usually consists of wide local resection, if possible, but a high local recurrence rate is common because of its location in
- 196. extracompartmental structures such as tendon sheaths. If that occurs, amputation is carried out for local control of the disease. Local radiation therapy is utilized with attempts at wide resection. Adjuvant chemotherapy has been advised because of the poor prognosis but the response is usually not beneficial.
- 197. CLASSIC Case #393 35 year female with clear cell sarcoma hand
- 198. Surgical exposure reveals extensive tendon sheath involvement
- 199. Photomic
- 200. Case#394 Sagittal gad contrast MRI tumor 73 year female with clear cell sarcoma ankle
- 201. tibia heel cord tumor Axial gad contrast MRI
- 202. Myositis Ossificans
- 203. Myositis Ossificans Myositis ossificans is a heterotopic ossification within muscle fascial planes seen typically in young athletic individuals in their adolescence and early adult life. It occurs primarily in males and usually results from a significant injury to a muscle, such as a tearing of the quadriceps muscle which is the most common location for this problem. It is also seen in the gluteus maximus and the brachialis muscle at the elbow. The calcification is typically noted on x-ray three to four weeks after the injury. It tends to occur at the periphery of the damaged muscle and hematoma is usually seen in the central area. As the lesion matures the calcific rim around the damaged muscle will appear as fairly mature bone and the central area will remain radiolucent, giving the so-called zonal pattern that is almost diagnostic of traumatic myositis ossificans. This is the opposite of osteosarcoma of soft tissue that has the most dense portion of the calcifying lesion occurring centrally and the more lytic portion at the periphery of the lesion. Myositis
- 204. ossificans can also be seen in older patients with no history of trauma in which case the clinician becomes concerned about the possibility of a neoplasm such as a synovial sarcoma or soft tissue osteosarcoma. Histologically, the lesion will have the appearance of a healing fracture, including immature cartilage and bone formation, along with hematoma in the early stages. In rare cases, after a period of 25 or 30 years, these dormant lesions can reactivate and develop into an osteosarcoma. Treatment usually consists of rest until the lesion matures after six months, at which point the patient is usually asymptomatic. There is no reason to remove the lesion unless there is significant clinical disability related to stiffness of the adjacent joint. There is a hereditary congenital form of myositis ossificans referred to as myositis ossificans progressiva, or the newer term- inology is fibrodysplasia ossificans progressiva, that is typically seen in children under the age of ten years. It presents with a clinical picture of progressive fibroblastic proliferation and
- 205. subsequent calcification and ossification of subcutaneous fat, muscles, tendons, appeneuroses, and ligaments. This condition can be associated with symmetrical malformations of the digits with microdactyly of the thumbs and great toes, sometimes associated with a failure of segmentation of the digital bony structures. The condition usually presents between birth and the first six years of age. It is inherited as an autosomal dominant trait. Males and females are equally affected and the calcification in soft tissues is usually precipitated by a local injury to the soft tissue. It occurs typically in the musculature of the back, shoulder, paravertebral region and upper arms. Fusion of the tempromandibular joint can be seen. If the respiratory muscles are affected, death can result because of respiratory failure or pneumonia in early adult life. The prognosis for survival is very poor and most patients die within the first ten to fifteen years of life. Biopsy or trauma of the affected areas should be avoided because of new lesions that might develop. There is no effective treatment or this condition.
- 206. CLASSIC Case #395 12 year female with myositis ossificans medial thigh
- 207. fibrous tissue Cut surgical specimen showing mature bone at periphery
- 208. Photomic showing reactive bone at periphery of lesion
- 209. Less bone maturity toward center of lesion
- 210. Healing rhabdomyoblasts in center of lesion
- 211. Case #396 59 year male with early myositis ossificans ant thigh
- 212. Lateral view
- 213. Axial T-2 MRI
- 214. Axial T-2 MRI
- 215. Macro section of resected specimen
- 216. bone Reactive bone at periphery
- 217. Case #397 biopsy 22 year male with early myositis ossificans anterior thigh
- 218. bone fibrous Biopsy specimen with typical benign zonal pattern
- 219. Mature myositis ossificans 6 mos later
- 220. Case #398 2 mos later early 17 year male with myositis ossificans of thigh 2 mos apart
- 221. Case #399 24 year female myositis ossificans 2 months post injury
- 222. Axial T-1 MRI
- 223. Axial T-2 MRI
- 224. Coronal T-2 MRI
- 225. Biopsy specimen showing reactive bone
- 226. cartilage bone Close up of reactive bone and cartilage
- 227. Case #400 14 month male with myositis ossificans brachialis muscle from fall 2 weeks ago
- 228. Case #401 14 year male with myositis ossificans triceps muscle mature
- 229. Case #402 10 year old female with myositis ossificans of shoulder
- 230. Case #403 62 year female with old myositis ossificans teres major
- 231. Coronal T-1 MRI
- 232. Superficial biopsy specimen shows benign reactive bone
- 233. A deeper specimen reveals OGS arising from previous myositis ossificans
- 235. CLASSIC Case #404 22 year male with myositis ossificans progressiva
- 236. Heavy ossification of latissimus dorsi muscle
- 237. X-ray showing ossification of latissimus dorsi muscle
- 239. Ossification psoas m
- 240. Heterotopic ossification seen muscle biopsy
- 241. Microdactyly both great toes
- 242. X-ray of feet showing failure of segmentation great toes
- 243. Hands showing stiff thumbs and hypoplastic 5th digits
- 244. X-ray showing hypoplasia of 1st and 5th digits
- 245. Case #405 26 year male with myositis ossificans progressiva with spontaneous fusion scoliotic spine
- 246. Spontaneous extra-articular fusion left hip
- 248. Pigmented Villonodular Synovitis The etiology of pigmented villonodular synovitis (PVNS) remains controversial. It presents as an inflammatory synovial disease, usually involving only one joint, but histologically the disease presents with histiocytic proliferation in the subsynovial tissue that takes on the characteristics of a neoplastic condition similar to that of a giant cell tumor. PVNS occurs typically in the subsynovial tissue about major joints of the lower extremity in patients between the ages of 20 and 40 years. The knee joint is the most common site, followed next by the hip, ankle and foot. It is rare to see this disease in the upper extremity. The histiocytic proliferation in subsynovial tissues is similar to that seen in giant cell tumor of tendon sheathes in the hand and foot., The clinical picture in the knee joint is that of spontaneous swelling associated with pain and synovial hypertrophy. Hemarthrosis can result in massive swelling about the knee joint and can occasionally result
- 249. in juxta-articular erosion of bone, similar to what is seen in rheumatoid synovitis. Other clinical conditions with a similar presentation include hemophilia and coccydiomycosis. In fewer than 10 % of cases this condition will present as a localized focal mass in the suprapatellar pouch of the knee or high in the popliteal space posteriorly that can masquerade as a neoplastic condition such as a synovial sarcoma. Treatment for the more generalized synovial involvement of the knee joint or other lower extremity joints consists of a subtotal synovectomy that in many cases can be performed through an arthroscope. In more extensive cases an open procedure may be necessary. The recurrence rate is fairly high, in the range of 30%. In cases where multiple recurrences result, treatment with radiation therapy in the neighborhood of 1500-3000 centigray by external beam is used. Injectable isotopes have also been used for radiation treatment of recurrent cases. Secondary arthritic changes, especially in the knee joint, can occur as a late complication of this disease
- 250. and these changes could lead to a total joint replacement at the age of 50 or 60 years. On very rare occasion, this disease can convert to a neoplastic sarcoma with a high degree of giant cell activity. This is similar to the conversion of a giant cell tumor to a malignant sarcoma.
- 251. CLASSIC Case #406 36 year male with PVNS knee
- 252. Lateral x-ray of knee showing soft tissue swelling
- 253. AP x-ray showing early osteoarthritis
- 254. Sagittal T-1 MRI showing hypertrophic synovitis bone erosion
- 255. hemosiderin cyst laden Sagittal T-2 MRI erosion
- 256. Axial T-1 MRI showing hypertrophic synovitis
- 257. cyst Coronal T-2 MRI
- 258. Brown discoloration of hypertrophic synovitis at surgery
- 259. Photomic showing giant cell activity
- 260. Photomic showing hemosiderin laden macrophages
- 261. Surgical appearance following subtotal synovectomy
- 262. Case #407 Sagittal proton density MRI 41 year female with PVNS knee
- 263. fluid hemosiderin laden synovitis Axial GRE T-2 MRI
- 264. Surgical exposure of brown hypertrophic synovitis
- 265. Photomic showing giant cell activity
- 266. Case #407.1 38 year male with intermittent pain and swelling rt knee 2 years
- 267. Sag T-2 T-2 Gad
- 268. Axial T-2 Gad
- 269. Case #407.2 Recurrent PVNS 43 yr male with 5 scopes over 7 yrs for intermittent painful swelling about the left knee
- 270. Sag PD T-2 FS T-2 FS
- 271. Axial T-2
- 272. Cor T-1 T-2
- 273. Case #408 55 year female PVNS knee with large subchondral lytic granuloma
- 274. Coronal T-1 MRI
- 275. Case #409 38 year male with PVNS hip joint with large supra-acetabular granuloma
- 276. Diffuse synovial hypertrophic nodularity seen with arthrogram
- 277. Treatment in the 60’s with a cup arthoplasty and bone graft to the acetabular granuloma
- 278. Case #410 Sagittal proton density MRI 17 male with localized nodular form of PVNS
- 279. Sagittal T-2 MRI
- 280. Axial T-2 MRI
- 281. Surgical specimen
- 282. Photomic showing hemosiderin laden macrophages
- 283. Case #410.1 Sag T-1 PD Nodular PVNS 45 year female with knee pain for 5 months
- 284. Cor T-1 PD FS
- 285. Surgical excision
- 286. Case #410.2 Localized nodular form of PVNS 32 yr male with dull pain R knee for 5 months
- 287. Sag T-1 T-2 Gad
- 288. Axial T-1 T-2 Gad
- 289. Cor T-1 T-2 FS Gad
- 290. Case #411 Sagittal proton density MRI erosion 50 year male with PVNS knee with large popliteal mass
- 291. Sagittal T-2 MRI
- 292. Case #412 Sagittal T-1 MRI 17 year female with PVNS ankle
- 293. Sagittal T-2 MRI
- 294. Another sagittal T-2 cut
- 295. Synovectomy specimen
- 296. Photomic
- 297. Case #413 Sagittal T-1 MRI 39 year female with diffuse form of GCT tendon sheath
- 298. Axial T-1 MRI
- 299. tumor Axial T-2 MRI
- 300. Photomic
- 301. Case #414 67 year female with giant cell tumor of tendon sheath
- 303. Case #415 14 year female with GCT tendon sheath
- 304. X-ray showing bony erosion
- 306. Case #416 14 year female with xanthomatous variant of giant cell tumor of tendon sheath on index finger
- 307. Surgical resection
- 308. Photomic showing cholesterol laden foam cells
- 309. Case # 1249 20 year female with PVNS knee
- 310. Lateral view
- 311. R L Bone scan
- 312. Sagittal T-1 MRI
- 313. Coronal T-1 MRI
- 314. Coronal gad contrast MRI
- 315. Case #1250 PVNS DOA 68 year male with PVNS and DOA left knee
- 316. Lateral view showing subchondral erosions
- 317. Axial T-1 MRI with subpatellar synovial hypertrophy
- 318. Sagittal T-1 MRI showing extensive suprapatellar pouch involvement
- 319. Sagittal T-2 MRI showing fluid in suprapatellar pouch
- 320. Coronal T-2 MRI with dark signal hemosiderin laden synovitis and bright signal synovial fluid
- 321. Another coronal T-2 MRI Subchondral erosions
- 322. Case #1251 34 year male with PVNS knee Sagittal T-1 MRI
- 324. Another sagittal GRE T-2* MRI
- 325. Coronal GRE T-2* MRI
- 326. Case #1252 23 year male with PVNS suprapatellar pouch area Coronal T-1 MRI
- 327. Coronal T-2 MRI
- 328. Axial T-2 MRI
- 329. Case #1253 Sagittal T-1 MRI 19 year male with localized nodular form of PVNS knee
- 330. Coronal T-1 MRI
- 331. Coronal T-2 MRI
- 332. Case #1254 40 year female with PVNS hip joint
- 333. CT scan
- 334. Axial T-1 MRI
- 335. Axial T-2 MRI
- 336. Case #1255 32 year male with PVNS hip joint
- 337. Case #1256 40 year female with pigmented villonodular tenosynovitis flexor hallicus longus tendon sheath Sagittal T-1 MRI
- 338. Coronal T-1 MRI
- 339. Axial T-1 MRI
- 340. Axial T-2 MRI
- 341. hemosiderin histiocytes Photomic showing giant cell activity
- 342. Case #1256.1 12 year old male with tender lump under heel cord 4 months
- 343. Sag T-1 PD Gad
- 344. Axial T-1 T-2 Gad
- 345. Case #1257 8 year female with GCT tendon sheath 2nd toe
- 346. Sagittal T-1 MRI
- 347. Coronal T-1 MRI
- 348. Case #1257.1 GCT tendon sheath 53 yr female with tender and swollen 2nd toe for 1 yr
- 349. Cor T-1 T-2 FS Gad
- 350. Axial T-1 T-2 FS Gad
- 351. Sag T-1 T-2 Gad
- 352. Intramuscular Myxoma
- 353. Case #1258 Axial T-2 Axial Gad 46 yr female with painless lump in post axilla for 8 years
- 354. Sagittal T-2 Sagittal Gad
- 355. Coronal Gad Surgical specimen
- 356. Case #1258.1 68 year old male with painless lump right shoulder for 1 yr
- 357. Cor T-1 T-2
- 358. Sag T-1 T-2
- 359. Case #1258.2 Axial T-1 T-2 59 year male with painless soft tissue mass medial calf for 6 mos. Gad Intramuscular myxoma
- 360. Sagittal T-1 T-2 Gad
- 361. Ganglion Cyst
- 362. Case #1259 Axial T-1 48 yr male with painless lump in anterior thigh 1 yr T-2
- 363. Axial Gad Shows rim enhancement Sagittal Gad
- 364. Case #1260 Ganglion cyst 49 year female with tender lump medial knee for 6 mos
- 365. Cor T-1 Cor T-2
- 366. Case #1160.1 Ganglion cyst 65 year female with non tender mass over patellar ligament
- 367. Sag T-1 PD FS
- 368. Cystic lateral meniscus Case #1261 Axial T-1 T-2 49 year male with prior history of subcutaneous ganglion cyst removed from knee area 6 years ago
- 369. Cor T-1 T-2
- 370. Case #1261.1 Axial T-1 T-2 Gad 72 year male with painless lump lateral ankle for 1 year
- 371. Cor STIR Surgical excision
- 372. Ganglion cyst Case #1261.2 Axial T-2 45 year male runner With tender nodule in Medial gastroc head Gad
- 373. Cor T-2 Gad
- 374. Case #1261.3 Ganglion cyst 60 year male with mild R hip pain for 1 year
- 375. Cor T-1 PD Gad
- 376. Axial T-2
- 377. Soft Tissue Lymphoma
- 378. Case #1262 Axial T-1 T-2 Lymphoma 100 year old male with chronic edema in legs but recent tender lump in calf for 3 months Gad
- 379. Sag T-1 STIR Gad