6. What is annotation? Who is involved? Term confusion (what’s in a name?) Scale: the sea of data Controlled vocabularies & ontologies The Gene Ontology Consortium Background: the language of biology 4
7. Annotation 5 annotate – to make or furnish critical or explanatory notes or comment. (Merriam-Webster dictionary) genome annotation – the process of taking the raw DNA sequence produced by the genome-sequencing projects and adding the layers of analysis and interpretation necessary to extract its biological significance and place it into the context of our understanding of biological processes. (Lincoln Stein, PMID 11433356) Gene Ontology annotation – the process of assigning GO terms to gene products… according to two general principles: first, annotations should be attributed to a source; second, each annotation should indicate the evidence on which it is based. (http://www.geneontology.org)
8. Diverse parties involved 6 End-users, including various researchers Small-scale laboratory projects Whole genome sequencing projects Annotators From reading papers to computational analysis Ontology developers Create terms that reflect scientific knowledge Make interoperable ontologies, database links Developers of tools & resources Standards for storing & sharing data Web interfaces for data analysis & sharing Many areas of expertise Laboratory sciences – biology, chemistry, medicine, and many other disciplines Computational science – bioinformatics, genomics, statistics Software development & web design Philosophy – ontology & logic
9. Term confusion: synonyms 7 Do biologists use precise & consistent language? Mutually understood concepts – DNA, RNA, or protein Synonym (one thing known by more than one name) – translation and protein synthesis Enzyme Commission reactions Standardized id, official name & alternative names http://www.expasy.ch/enzyme/2.7.1.40
11. Term confusion: homonyms and biological complexity 9 AmiGO query “vascular” 51 terms In biology, many related phenomena are described with similar terminology
20. How to draw meaningful comparisons?http://en.wikipedia.org/wiki/File:Microarray2.gif (accessed 17-Sep-09)
21. The Gene Ontology (GO) 11 Way to address the problems of synonyms, homonyms, biological complexity, increasing glut of data GO provides a common biological language for protein functional annotation www.geneontology.org
22. Controlled vocabulary (CV) 12 An official list of precisely defined terms that can be used to classify information and facilitate its retrieval Think of flat list like a thesaurus or catalog Benefits of CVs Allow standardized descriptions of things Remedy synonym & homonym issues Can be cross-referenced externally Facilitate electronic searching A CV can be “…used to index and retrieve a body of literature in a bibliographic, factual, or other database. An example is the MeSH controlled vocabulary used in MEDLINE and other MEDLARS databases of the NLM.” http://www.nlm.nih.gov/nichsr/hta101/ta101014.html
23. Ontology is a type of CV with defined relationships 13 Ontology – formalizes knowledge of a subject with precise textual definitions Networked terms where child more specific (“granular”) than parent Less specific GO terms describe biological attributes of gene products… More granular
24. How GO works 14 GO Consortium develops & maintains: Ontologies and cross-links between ontologies and different resources Tools to develop and use the ontologies SourceForge tracker for development People studying organisms at databases annotate gene products with GO terms Groups share files of annotation data about their respective organisms Because a common language was used to describe gene products and this information was shared amongst databases… We can search uniformly across databases Do comparative genomics of diverse taxa
26. The Gene Ontology Consortium 16 Collaboration began 1998 among model organism databases mouse (MGI), fruit fly (FlyBase) and baker’s yeast (SGD) Michael Ashburner of FlyBase contributed the base vocabulary Today > 20 members & associates First publication 2000 (PMID 10802651) Today, PubMed query “gene ontology” yields 3,347 papers (27-Jun-2011) Organisms represented by GO annotations from every kingdom of life Many groups use GO in many different ways for their research Among eight OBO-Foundry ontologies ZFIN Reactome IGS
27. OBO Foundry ontologieswww.obofoundry.org 17 Collaboration among developers of science-based ontologies Establish principles for ontology development Goal of creating a suite of orthogonal interoperable reference ontologies in the biomedical domain. many others…
28. What the GO is not GO comprises three ontologies Anatomy & storage of GO terms Ontology structure Detail of a term in AmiGO True path rule Gene Ontology:overview, terms & structure 18
29. Caveats – what GO is not 19 Not gene naming system or gene catalog GO describes attributes of biological objects – “oxidoreductase activity” not “cytochromec” The three ontologies have limitations No sequence attributes or structural features No characteristics unique to mutants or disease No environment, evolution or expression No anatomy features above cellular component Not dictated standard or federated solution Databases share annotations as they see fit Curators evaluate differently GO is evolving as our knowledge evolves New terms added on daily basis Incorrect/poorly defined terms made obsolete Secondary ids – terms with same meaning merged
30. GO comprises three ontologies 20 Cellular component ontology (CC) “cytoplasm” Molecular function ontology (MF) “protein binding” “peptidase activity” “cysteine-type endopeptidase activity” Biological process ontology (BP) “proteolysis” “apoptosis” Terms describe attributes of gene products (GPs) Any protein or RNA encoded by a gene Species-independent context, e.g. “ribosome” Could describe GPs found in limited taxa, e.g. “photosynthesis” or “lactation” One GP can be associated with ≥ 1 CC, BP, MF Example: Caspase-6 from Bostaurus
53. Anatomy of a GO term 24 Term name goid (unique numerical identifier) Synonyms (broad or narrow) for searching, alternative names, misspellings… Precise textual definition with reference stating source GO slim Ontology placement
60. GO has three term relationships 28 is_a - child is instance of parent (“A is_a B”) Class-subclass relationship part_of - child part of parent (“C part_of D”) When C present, part of D; but C not always present Nucleus always part_of cell; not all cells have nuclei regulates Child term regulates parent term (Zoomed in view of biological process ontology depicted here.)
61. AmiGO for viewing terms 29 Open source HTML-based application developed by the GO Consortium Interface for browsing, querying and visualizing OBO data Users can search GO terms or annotations Available via website or download for local install http://amigo.geneontology.org Example query with keyword “hemolysis” or goid GO:0019836 GO:0019836
64. AmiGO view continued 32 Several informative views Click Number of gene products in GO annotation collection annotated to that term or one of its child terms Relationship between term and its parent Our term is much further down…
67. Definition: Catalysis of the transfer of amino acids from one side of a membrane to the other. Amino acids are organic molecules that contain an amino group and a carboxyl group. [source: GOC:ai, GOC:mtg_transport, ISBN:0815340729]
72. “True path rule” 36 The pathway from a term all the way up to its top-level parent(s) must always be true for any gene product that could be annotated to that term (“if true for the child, then true for the parent”) Incorrect for Bacteria cell organelle mitochondrion proton-transporting ATP synthase complex Correct for Bacteria (and Eukaryotes) cell intracellular proton-transporting ATP synthase complex plasma membrane proton-transporting ATP synthase complex mitochondrial proton-transporting ATP synthase complex membrane plasma membrane plasma membrane proton-transporting ATP synthase complex organelle mitochondrion mitochondrial inner membrane mitochondrial proton-transporting ATP synthase complex (Abbreviated versions of the actualtrees)
73. What is GO annotation? Literature curation at model organism databases The annotation file Evidence – critical for annotation Sequence similarity-based annotation Annotation specificity Annotating with GO and Evidence 37
74. GO annotation overview 38 Associating a GO term with a gene product Goal is to select GO terms from all three ontologies to represent what, where, and how Linking a GO term to a gene product asserts that it has that attribute For example, 6-phosphofructokinase Molecular function GO:0003872 6-phosphofructokinase activity Biological process GO:0006096 glycolysis Cellular component GO:0005737 cytoplasm Annotation, whether based on literature or computational methods, always involves: Learning something about a gene product Selecting an appropriate GO term Providing an appropriate evidence code Citing a [preferably open access] reference Entering information into GO annotation file
75. Chaperone DnaK, one protein/multiple annotations 39 Molecular function ATP binding (GO:0005524) ATPase activity (GO:0016887) unfolded protein binding (GO:0051082) misfolded protein binding (GO:0051787) denatured protein binding (GO:0031249) Biological process protein folding (GO:0006457) protein refolding (GO:0042026) protein stabilization (GO:0050821) response to stress (GO:0006950) Cellular component cytoplasm (GO:0005737)
77. Results section indicates a “direct assay” annotation 41 They document the findings of a direct assay performed on purified protein: They further document the methods used, and evaluate the findings in the Discussion section…
78. Query AmiGO with “DNA ligase” & “DNA ligation” 42 All “ligation” in biological process ontology
79. Resulting annotations 43 Name: DNA ligase (stated in paper) Gene symbol: ligA (stated in paper) EC: 6.5.1.2 (queried enzyme for “DNA ligase”)
81. Evidence 45 Essential to base annotation on evidence Conclusions more robust and traceable With evidence, a GO annotation is standard operating procedure (SOP)-independent Many types of evidence exist For example, experiment described in literature What method (e.g. direct assay, mutant phenotype, et cetera) was used? Did author cite references? Did author provide details of analyses? Perhaps you used a sequence-based method What were the methods of manual curation? Give accession numbers of similar sequences Provide any references describing methods Controlled vocabularies help here, too!
82. GO standard references 46 GO_REF:0000011 A Hidden Markov Model (HMM) is a statistical representation of patterns found in a data set. When using HMMs with proteins, the HMM is a statistical model of the patterns of the amino acids found in a multiple alignment of a set of proteins called the "seed". Seed proteins are chosen based on sequence similarity to each other. Seed members can be chosen with different levels of relationship to each other... GO_REF:0000011 A Hidden Markov Model (HMM) is a statistical representation of patterns found in a data set. When using HMMs with proteins, the HMM is a statistical model of the patterns of the amino acids found in a multiple alignment of a set of proteins called the "seed". Seed proteins are chosen based on sequence similarity to each other. Seed members can be chosen with different levels of relationship to each other. They can be members of a superfamily (ex. ABC transporter, ATP-binding proteins), they can all share the same exact specific function (ex. biotin synthase) or they could share another type of relationship of intermediate specificity (ex. subfamily, domain). New proteins can be scored against the model generated from the seed according to how closely the patterns of amino acids in the new proteins match those in the seed. There are two scores assigned to the HMM which allow annotators to judge how well any new protein scores to the model. Proteins scoring above the "trusted cutoff" score can be assumed to be part of the group defined by the seed. Proteins scoring below the "noise cutoff" score can be assumed to NOT be a part of the group. Proteins scoring between the trusted and noise cutoffs may be part of the group but may not. One of the important features of HMMs is that they are built from a multiple alignment of protein sequences, not a pairwise alignment. This is significant, since shared similarity between many proteins is much more likely to indicate shared functional relationship than sequence similarity between just two proteins. The usefulness of an HMM is directly related to the amount of care that is taken in chosing the seed members, building a good multiple alignment of the seed members, assessing the level of specificity of the model, and choosing the cutoff scores correctly. In order to properly assess what functional relevance an above-trusted scoring HMM match has to a query, one must carefully determine what the functional scope of the HMM is. If the HMM models proteins that all share the same function then it is likely possible to assign a specific function to high-scoring match proteins based on the HMM. If the HMM models proteins that have a wide variety of functions, then it will not be possible to assign a specific function to the query based on the HMM match, however, depending on the nature of the HMM in question, it may be possible to assign a more general (family or subfamily level) function. In order to determine the functional scope of an HMM, one must carefully read the documentation associated with the HMM. The annotator must also consider whether the function attributed to the proteins in the HMM makes sense for the query based on what is known about the organism in which the query protein resides and in light of any other information that might be available about the query protein. After carefully considering all of these issues the annotator makes an annotation.
83. GO evidence codeswww.geneontology.org/GO.evidence.shtml 47 EXP - inferred from experiment IDA - inferred from direct assay IEP inferred from expression pattern IGI - inferred from genetic interaction IPI - inferred from physical interaction IMP - inferred from mutant phenotype ISS - inferred from sequence or structural similarity ISA - inferred from sequence alignment ISO - inferred from sequence orthology ISM - inferred from sequence model IGC - inferred from genomic context ND - no biological data available IC - inferred by curator TAS - traceable author statement NAS - non-traceable author statement IEA - inferred from electronic annotation GO codes are a subset of yet another ontology!
84. Types of sequence similarity-based annotations 48 Find similarity between gene product & one that is experimentally characterized BLAST-type alignments Shared synteny to establish orthology of genomic regions between species Find similarity between gene product and defined protein family HMMs (Pfam, TIGRFAMS) Prosite InterPro Find motifs in gene product with prediction tools TMHMM SignalP Many (most?) information you find is based on transitive annotation and much of it has never been looked at by a human being!
85. Evaluation of sequence similarity-based information 49 Visually inspect alignments & criteria Length & identity Conservation of catalytic sites Check HMM scores with respect to cutoff Look at available metabolic analysis Pathways, complexes? Information from neighboring genes Gene in an operon (common prokaryotes) can supplement weak similarity evidence Sequence characteristics Transmembraneregions? Signal peptide? Known motifs that give a clue to function? Paralogous family member
86. An example: HI0678, a protein from H. influenzae… ...high quality alignment to experimentally characterized triosephosphateisomerase from Vibrio marinus 50
88. High quality….. …. full-length match, high percent identity (67.8%), conserved active and binding sites (boxed in red). 52
89. Resulting annotations 53 name:triosephosphateisomerase gene symbol:tpiA EC: 5.3.1.1 (This, and the following annotations, came from the match protein.)
93. And another annotation 57 The biologist knows that glycolysis takes place in the cytoplasm in bacteria, and so infers a cytoplasmic location for that protein (“inferred by curator” evidence code).
94. Annotation specificity should reflect knowledge 58 GO trees (very abbreviated) Function catalytic activity kinase activity carbohydrate kinase activity ribokinase activity glucokinase activity fructokinase activity Process metabolism carbohydrate metabolism monosaccharide metabolism hexose metabolism glucose metabolism fructose metabolism pentose metabolism ribose metabolism Available evidence for three genes #1 -good match to an HMM for “kinase” #2 -good match to an HMM for “kinase” -a high-quality BER match to an experimentally characterized “glucokinase’ AND a ‘fructokinase’ #3 -good match to an HMM specific for “ribokinase” -a high-quality BER match to an experimentally characterized ribokinase #1 #2 #3 #1 #2 #3
95. Using shared annotations Search for GO terms at databases Slims for broad classification GO tools Working with GO-limited data sets Summary Using annotation to facilitate your research 59
96. Sharing annotations 60 Annotation file sent to GO, put in repository All these data free to anyone Hundreds of thousands of GP annotations Annotation files all in same format Facilitates easy use of data by everyone Most of your favorite organism databases use these annotation files
98. 62 Ontology slim www.geneontology.org/GO.slims.shtml Slim is a distilled (reduced) ontology Made by manually pruning low-level terms with an ontology editor Selected high-level terms remain Slims reduce ontology complexity Reduce clutter & see general trends Microarray experiments Comparative whole genome analyses Remove irrelevant terms Looking at specific taxa, such as yeast or plant Go offers script to bin more granular annotations up to higher levels
101. GO toolswww.geneontology.org/GO.tools.shtml 65 The real challenge is finding the right one for your needs For example, statistical representation of GO terms: http://go.princeton.edu/cgi-bin/GOTermFinder
102. GO & analysis of RNA-seqdata 66 Young et al. Genome Biology 2010, 11:R14 http://genomebiology.com/2010/11/2/R14 We present GOseq, an application for performing Gene Ontology (GO) analysis on RNA-seq data. GO analysis is widely used to reduce complexity and highlight biological processes in genome-wide expression studies, but standard methods give biased results on RNA-seq data due to over-detection of differential expression for long and highly expressed transcripts. Application of GOseq to a prostate cancer data set shows that GOseq dramatically changes the results, highlighting categories more consistent with the known biology.
103. When GO is limited 67 Food for thought: what happens when we have limited GO (or other)annotation data? New and interesting genomes often see this problem
104. Comparative analysis of orthologs in syntenic blocks 68 The more genomes we have at our disposal, the better Structural rearrangements, absence of intron, gene duplication, intron structure, gene deletion/creation Nucleic Acids Res. 2010 January; 38(Database issue): D420–D427.
105. Summary GO analyses 69 GO remedies problems of synonyms & homonyms in biological nomenclature Queries based on IDs linked to precise definitions, not less reliable text-matching GO can help you to: Find all genes that share a particular function regardless of sequence Do comparisons across any species annotated with GO Summarize major classes of genes in a newly sequenced genome Characterize expressed genes is a study Drive hypotheses to test in the laboratory GO is not a panacea but it should be a valuable tool in your genomics toolbox
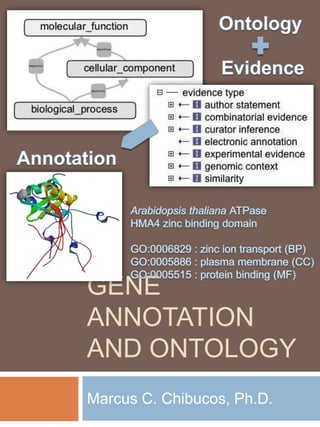
106. The title slide revisited… Ontology Evidence Annotation Arabidopsis thaliana ATPase HMA4 zinc binding domain GO:0006829 : zinc ion transport (BP) GO:0005886 : plasma membrane (CC) GO:0005515 : protein binding (MF) Thank you.
Editor's Notes
In this report, we describe the cloning and expression of a Deinococcusradiodurans DNA ligase in Escherichia coli. This enzyme efficiently catalyses DNA ligation in the presence of Mn(II) and NAD+ as cofactors…
![Outline of this talk 2 ,[object Object]](data:image/gif;base64,R0lGODlhAQABAIAAAAAAAP///yH5BAEAAAAALAAAAAABAAEAAAIBRAA7)