
Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Similar a Hemoglobinopathy & erythrocyte enzyme disorder
Similar a Hemoglobinopathy & erythrocyte enzyme disorder (20)
Más de Marudhar Kesari Jain College for Women Vaniyambadi - 635 751, Tamil Nadu, INDIA.
Más de Marudhar Kesari Jain College for Women Vaniyambadi - 635 751, Tamil Nadu, INDIA. (20)
Último
Último (20)
Hemoglobinopathy & erythrocyte enzyme disorder
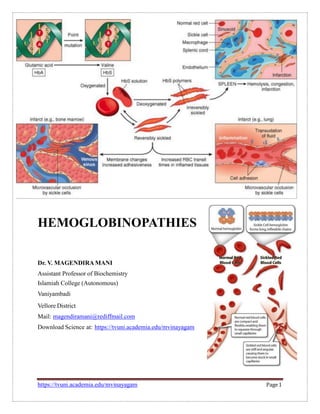
- 1. HEMOGLOBINOPATHIES Dr. V. MAGENDIRA MANI Assistant Professor of Biochemistry Islamiah College (Autonomous) Vaniyambadi Vellore District Mail: magendiramani@rediffmail.com Download Science at: https://tvuni.academia.edu/mvinayagam https://tvuni.academia.edu/mvinayagam Page1
- 2. Structure of Hemoglobin Hemoglobin is the protein molecule in red blood cells that carries oxygen from the lungs to the body's tissues and returns carbon dioxide from the tissues back to the lungs. Hemoglobin is made up of four protein molecules (globulin chains) that are connected together. A low hemoglobin count is generally defined as less than 13.5 grams of hemoglobin per deciliter (135 grams per liter) of blood for men and less than 12 grams per deciliter (120 grams per liter) for women. Approximately 6.25 gm of Hb are synthesized and destroyed every day. Heme is iron porphyrin compound. Porphyrin is a tetrapyrrole structure. Ferrous iron occupies the center of the porphyrin ring and establishes linkages with all the four nitrogens of all the pyrrole rings. It is also linked to nitrogen of imidazole ring of histidine present in globin part. Globin part is made of four polypeptide chains, to identical α-chains and two identical β-chains in normal adult hemoglobin. Heme consists of a porphyrin, called protoporphyrin IX, with a ferrous iron chelated in its center. Protoporphyrin IX contains four five-member, nitrogen containing pyrrole rings, held together by methane (-CH=) bridges and decorated with methyl (-CH3), vinyl (—CH=CH2), and propionate (— CH2—CH2—COO–) side chains. The most important part of the heme group is its iron. The iron in heme is bound to the nitrogen atoms of the four pyrrole rings. In hemoglobin and myoglobin, the iron forms a fifth bond with a nitrogen atom in a histidine side chain of the apoprotein. This histidine is called the proximal histidine. An optional sixth bond can be formed with molecular oxygen. Iron can exist in a ferrous (Fe2+) and a ferric (Fe3+) state. The heme iron in hemoglobin and myoglobin is always in the ferrous state. Hemoglobin has four polypeptides, each with its own heme. Humans have several types of hemoglobin. https://tvuni.academia.edu/mvinayagam Page2
- 3. Hemoglobin A (HbA), which contains two α-chains and two β-chains, is the major adult hemoglobin. The minor adult hemoglobin (HbA2) and fetal hemoglobin (HbF) also have two α- chains, but instead of the β-chains, HbA2 has δ-chains and HbF has γ -chains. The α-chains have 141 amino acids, and the β-, γ -, and δ -chains have 146 amino acids. All of these chains are structurally related. Varieties of normal human Hb are Hb-A1 (two α-chains and β-chains) HbF (two α-chains and γ -chains) Hb-A2 (two α-chains and delta-chains) Embyonic Hb (two α-chains and €-chains) Hb-A3 (Altered from Hb-A found in old red cells) HbA1C (Glycosylated Hb, present in concentration of 3-5% of total Hb). In diabetes mellitus it is increased to 6 to 15%. Abnormal Haemoglobins: More than 30 abnormal types descries, differentiated by their characteristic electrophoretic mobilities, generally transmitted; are due to single mutant gene; Two types https://tvuni.academia.edu/mvinayagam Page3
- 4. Due to mutation of structural gene. E.g: HbS, HbM, HbC, HbD (Punjab) etc. Due to mutation in regulator gene. E.g: Thalassemias. Detection by Finger Printing techniques and Hybridization Effects of abnormal Hb: Changed Red cell morphology Haemolytic anemia, Jaundice Methamoglobinemia High O2 affinity E.g: Hb chesapeake, Hb-Rainier Interfere with mRNA formation e.g.: Hb constant spring Normal Hemoglobin’s HemoglobinA This is the designation for the normal hemoglobin that exists after birth. Hemoglobin A is a tetramer with two alpha chains and two beta chains (α2β2). HemoglobinA2 This is a minor component of the hemoglobin found in red cells after birth and consists of two alpha chains and two delta chains (α2δ2). Hemoglobin A2 generally comprises less than 3% of the total red cell hemoglobin. HemoglobinA3 This is a minor component of the older form of hemoglobin found in old red cells. Hemoglobin A3 generally comprises less than 3 – 10 % of the total red cell hemoglobin. Embryonic Hb This is a minor component of the hemoglobin found in red cells three months after birth and consists of two alpha chains and two epsilon chains (α2ε2). Hemoglobin F Hemoglobin F is the predominant hemoglobin during fetal development. The molecule is a tetramer of two alpha chains and two gamma chains (α2γ2). The genes for hemoglobin F and https://tvuni.academia.edu/mvinayagam Page4
- 5. hemoglobin A are closely related, existing in the same gene cluster on chromosome 11. Hemoglobin F production falls dramatically after birth, although some people continue to produce small amounts of hemoglobin F for their entire lives. Glycosylated Hb (HbA1c) This is a minor component of the hemoglobin found in red cells. HbA1c generally comprises less than 3 – 5 % of the total red cell hemoglobin. During diabetes mellitus the range may exceed 6-15 % this gives index of blood sugar. 6–7% (42–53 mmol/mol Hb) taken to indicate good diabetic control. The concentration of HbA1c is dependent on the concentration of glucose in the blood and the duration of hyperglycemia. In prolonged hyperglycemia the concentration may rise to 12% or more of the total hemoglobin. Patients with diabetes mellitus have high concentrations of blood glucose and therefore high amounts of HbA1c. The changes in the concentration of HbA1c in diabetic patients can be used to follow the effectiveness of treatment for the diabetes. HEMOGLOBINOPATHY Hemoglobinopathy is a kind of genetic defect that results in abnormal structure of one of the globin chains of the hemoglobin molecule. Hemoglobinopathies are inherited single-gene disorders; in most cases, they are inherited as autosomal co-dominant traits. Common hemoglobinopathies include sickle-cell disease, Thalassemias. Hemoglobinopathies denote structural abnormalities in the globin proteins (hemoglobinopathy) or underproduction of normal globin proteins (thalassemia), often through mutations in regulatory genes. Either hemoglobinopathy or thalassemia, or both, may cause anemia. Some well-known hemoglobin variants such as sickle-cell anemia and congenital dys-erythropoietic anemia are responsible for diseases, and are considered hemoglobinopathies. Hemoglobin is produced by genes that control the expression of the hemoglobin protein. Defects in these genes can produce abnormal hemoglobins and anemia, which are conditions termed "hemoglobinopathies". Abnormal hemoglobins appear in one of three basic circumstances: https://tvuni.academia.edu/mvinayagam Page5
- 6. Structural defects in the hemoglobin molecule Alterations in the gene for one of the two hemoglobin subunit chains, alpha (α) or beta (β), are called mutations. Often, mutations change a single amino acid building block in the subunit. Most commonly the change is harmless or disturbing neither the structure nor function of the hemoglobin molecule. Occasionally, alteration of a single amino acid dramatically disturbs the behavior of the hemoglobin molecule and produces a disease state. Sickle hemoglobin exemplifies this phenomenon. Diminished production of one of the two subunits of the hemoglobin molecule Mutations that produce this condition are termed "thalassemias." Equal numbers of hemoglobin alpha and beta chains are necessary for normal function. Hemoglobin chain imbalance damages and destroys red cells thereby producing anemia. Although there is a shortage of the affected hemoglobin subunit, with most thalassemias the few subunits synthesized are structurally normal. Abnormal associations of otherwise normal subunits A single subunit of the alpha chain (from the α-globin locus) and a single subunit from the β- globin locus combine to produce a normal hemoglobin dimer. With severe α-thalassemia, the β- globin subunits begin to associate into groups of four (tetramers) due to the lack of potential α- chain partners. These tetramers of β-globin subunits are functionally inactive and do not transport oxygen. ABNORMALHb’s Clinically Significant Variant Hemoglobin’s Hemoglobin S In both β-chains glutamic acid in 6th position is replaced by Valine (α2β2 A6Val ). This results in increase of viscosity and precipitation of HbS. Hence the crescent or sickle shaped RBC of more fragile nature. However such RBCs show increased resistance to malaria, but more vulnerable to salmonella infections. https://tvuni.academia.edu/mvinayagam Page6
- 7. Hemoglobin C In both β-chains glutamic acid in 6th position is replaced by Lysine (α2 A β2 6lys ). Hemoglobin C disease is relatively benign, producing a mild hemolytic anemia and splenomegaly. Hemoglobin C trait is benign. Hemoglobin D In both β-chains glutamic acid in 121th position is replaced by Glutamine (α2 A β2 121Gln ). Hemoglobin E In both β-chains glutamic acid in 26th position is replaced by Lysine (α2 A β2 26Lys ). This variant results from a mutation in the hemoglobin beta chain. People with hemoglobin E disease have a mild hemolytic anemia and mild splenomegaly. Hemoglobin E trait is benign. Hemoglobin M In this one amino acid sequence is altered either in alpha and beta chains. Different types of Hb M. Hb-M (Iwate) - histidine of position 87 of alpha chain has been replaced by Tyrosine (α2 ATyr β2 A ). The oxygen affinity of Hb-M (Iwate) is much lower than that of normal Hb-A. Hemoglobin Sabine leucine of position 91 of alpha chain has been replaced by prolineHb-M (Sabine) - (α2 A β2 91Pro ). Hemoglobin Chesapeake Lysine of position 92 of alpha chain has been replaced by Arginine (α2 92Arg β2 A ). Leads to decreased oxygen affinity, resulting tissue hypoxia, polycythemia. Hemoglobin Rainier Tyrosine of position 145 of alpha chain has been replaced by Histidine (α2 92Arg β2 A ). Leads to decreased oxygen affinity, resulting tissue hypoxia, polycythemia. https://tvuni.academia.edu/mvinayagam Page7
- 8. Hemoglobin Constant Spring It produces an unstable m-RNA. In this the ―UAA‖ stop codon has mutated to a ―CAA‖ which codes for Glutamine. Hence the alpha chain are 31 residues longer than normal (in place of 141 amino acids it has 172 amino acids). The longer m-RNA is instable and gets degraded readily. SICKLE CELLANAEMIA Due to a single nucleotide substitution (adenine to thymine) in the codon for amino acid 6 of globin; this change converts a glutamic acid codon (GAG) to a valine codon (GTG). This form of haemoglobin is referred to as HbS; normal adult haemoglobin is referred to as HbA. Substitution of a hydrophobic (valine) for a polar residue (glutamic acid) results in haemoglobin tetramers that aggregate upon deoxygenation in the tissues. Aggregation results in deformation of the red blood cell into a sickle-like shape, making it relatively inflexible and unable to easily traverse the capillary beds. Sickle cell anaemia is an autosomal recessive disorder. Individuals who are heterozygous are said to have a sickle cell trait. Although heterozygous individuals are clinically normal, their red blood cells can ‗sickle‘ under very low oxygen pressure, for example at high altitudes. Heterozygous individuals exhibit phenotypic dominance, yet are recessive genotypically. THALASSEMIAS The thalassemias are hereditary hemolytic diseases in which an imbalance occurs in the synthesis of globin chains. As a group, they are the most common single gene disorders in humans. Normally, synthesis of the α- and β-globin chains is coordinated, so that each α-globin chain has a β-globin chain partner. This leads to the formation of α2β2 (Hb A). In the thalassemias, the synthesis of either the α- or the β-globin chain is defective. A thalassemia can be caused by a variety of mutations, including entire gene deletions, or substitutions or deletions of one to many nucleo tides in the DNA. Each thalassemia can be classified as either a disorder in which no globin chains are produced (αo - or βo -thal assemia), or one in which some chains are synthesized, but at a reduced level (α+- or β+-thalassemia). https://tvuni.academia.edu/mvinayagam Page8
- 9. α-Thalassemias These are defects in which the synthesis of α-globin chains is decreased or absent, typically as a result of deletional mutations. Because each individual‘s genome contains four copies of the α- globin gene (two on each chromosome 16), there are several levels of α-globin chain 1deficiencies. If one of the four genes is defective, the individual is termed a silent carrier of α- thalassemia, because no physical manifestations of the disease occur. If two α-globin genes are defective, the individual is designated as having α -thalassemia trait. If three α-globin genes are defective, the individual has Hb H (β4) disease—a mildly to moderately severe hemolytic anemia. If all four α-globin genes are defective, Hb Bart (γ4) disease with hydrops fetalis and fetal death results, because α-globin chains are required for the synthesis of Hb F. Hemoglobin H Hemoglobin H is a tetramer composed of four beta globin chains. Hemoglobin H occurs only with extreme limitation of alpha chain availability. Hemoglobin Barts Hemoglobin Barts develops in fetuses with four-gene deletion alpha thalassemia. During normal embryonic development, the episilon gene of the alpha globin gene locus combines with genes from the beta globin locus to form functional hemoglobin molecules. The episolon gene turns off at about 12 weeks, and normally the alpha gene takes over. With four-gene deletion alpha thalassemia no alpha chain is produced. The gamma chains produced during fetal development combine to form gamma chain tetramers. These molecules transport oxygen poorly. Most individuals with four-gene deletion thalassemia and consequent hemoglobin Barts die in utero (hydrops fetalis). Hemoglobin Portland In this episilon chains and gamma chains are produced in excess and can form γ2 ε2. Foetuses survive for some time by making increased amounts of embryonic Hb, but they commonly die before term or shortly after delivery (hydrops fetalis). https://tvuni.academia.edu/mvinayagam Page9
- 10. β-Thalassemias In these disorders, synthesis of β-globin chains is decreased or absent, typically as a result of point mutations that affect the production of functional mRNA; however, α-globin chain synthesis is normal. α-Globin chains cannot form stable tetramers and, therefore, precipitate, causing the premature death of cells initially destined to become mature RBCs. Increase in α2γ2 (Hb F) and α2δ2 (Hb A2) also occurs. There are only two copies of the β-globin gene in each cell (one on each chromosome Therefore, 11). individuals with β-globin gene defects have either β-thalassemia trait (β-thalassemia minor) if they have only one defective β-globin gene, or β-thalassemia major (Cooley anemia) if both genes are defective. Hemoglobin partland Compound Heterozygous Conditions or Hemoglobin SC disease Hemoglobin is made of two subunits derived from genes in the alpha gene cluster on chromosome 16 and two subunits derived from genes in the beta gene cluster on chromosome 11. Occasionally someone inherits two different variant genes from the alpha globin gene cluster or two different variant genes from the beta globin gene cluster (a gene for hemoglobin S and one https://tvuni.academia.edu/mvinayagam Page10
- 11. for hemoglobin C, for instance). This condition is called "compound heterozygous". The nature of two genes inherited determines whether a clinically significant disease state develops. The compound heterozyous states tends to consist of common groupings (e.g., hemoglobin SC), due to the geographic clustering of hemoglobin variants around the world. ERYTHROCYTE ENZYME DISORDER The mature red blood cell (RBC), is optimally adapted to perform its most important function during its estimated 120-day lifespan in the circulation: the binding, transport and delivery of oxygen to all tissues. For this, the RBC requires three essential metabolic pathways: Anaerobic glycolysis, which is the only source of energy (ATP production) for maintenance of cell structure and function. Anti-oxidant pathways necessary for the protection of RBC proteins against oxidation, through the synthesis of glutathione (GSH), and of haemoglobin against iron oxidation through the maintenance of iron in its functional, reduced, ferrous state (cytochrome b5 reductase). https://tvuni.academia.edu/mvinayagam Page11
- 12. Nucleotide metabolism for the maintenance of the purine and pyrimidine nucleotides. In general, these enzymopathies are associated, in addition to haemolytic anaemia (acute or chronic), with systemic (non-haematological) manifestations such as neuropathy (with or without mental retardation), muscular disease, recurrent infections and metabolic acidosis. GLUCOSE-6-PHOSPHATE DEHYDROGENASE (G6PD) DEFICIENCY G6PD catalyzes the first step in the hexose monophosphate shunt which is necessary for producing NADPH. NADPH in turn is required for the maintenance of reduced glutathione (GSH), a tri- peptide that protects the RBC from oxidative damage. G6PD is distributed in all cells and the active enzyme is a monomer of 515 amino acids, with a molecular weight of about 59 kDa. The enzyme is active as a tetramer or dimmer, depending on pH. G6PD deficiency is the most common known enzymopathy and it is estimated to affect 400 million people worldwide. G6PD deficiency is X-linked and caused by different mutations in the G6PD gene, resulting in protein variants with different levels of enzyme activity that are associated with a wide range of biochemical and clinical phenotypes. Major clinical manifestations are Severe G6PD deficiency (1-5%) associated with acute haemolytic anaemia (AHA), (< 1%) chronic haemolytic anaemia (CHA) Severe anemia Haemoglobinuria (dark urine) Eccentrocytosis or bite-cells https://tvuni.academia.edu/mvinayagam Page12
- 13. Patients with CHA due to G6PD deficiency have, in general, a history of severe neonatal jaundice, chronic anaemia impaired by oxidative stress, blood transfusion requirement, splenomegaly and gallstones at early age. Chronic haemolysis attributable to G6PD deficiency is sometimes worsened by co-inherited (and unrelated) genetic erythrocyte alterations, such as membrane defects, thalassaemia, glucose-6-phosphate isomerase deficiency, pyruvate kinase deficiency and congenital dys-erythropoietic anaemia. Unexpectedly high amounts of non- conjugated bilirubin can be seen in the co-inheritance of G6PD deficiency and Gilbert‘s syndrome. PYRUVATE KINASE (PK) DEFICIENCY PK catalyzes the irreversible transfer of a phosphoryl group from phosphoenolpyruvate (PEP) to ADP, thus yielding pyruvate and ATP; it is a regulatory key enzyme of the glycolytic pathway. There are four iso-zymes of PK: PK-M1, PK-M2, PK-L, and PK-R. The PK-M1 iso-enzyme is expressed in skeletal muscle, heart, and brain, and it is the only isoenzyme that is not subject to allosterically regulation. PK-R is exclusively expressed in RBCs, whereas PK-L is predominantly expressed in the liver. The active PK-R enzyme is a tetramer of four identical subunits and each subunit is divided into 4 domains. The enzyme is allosterically activated by its substrate fructose-bis-phosphate (FBP) and negatively regulated by ATP. Furthermore, PK has an absolute requirement for cations Mg2+ and Ca+ . PK deficiency is the most common enzymopathy associated with CHA, and about 300 patients have been reported. Major clinical manifestations are Increased 2,3-BPG levels ameliorate the anaemia by lowering the oxygen-affinity of haemoglobin. Severe haemolysis causing neonatal death Very rare cases can present with hydrops foetalis (is a serious fetal condition defined as abnormal accumulation of fluid in two or more fetal compartments, including ascites, pleural effusion, pericardial effusion, and skin edema. In some patients, it may also be associated with polyhydramnios and placental edema.) https://tvuni.academia.edu/mvinayagam Page13
- 14. GLUCOSE PHOSPHATE ISOMERASE (GPI) DEFICIENCY GPI deficiency is the second most common erythro-enzymopathy of glycolytic enzymes after PK deficiency, and approximately 50 different cases have been described to date. GPI deficiency is an autosomal recessive genetic disorder associated with mild to severe CHA in homozygotes or compound heterozygotes. In a very few cases, GPI deficiency is associated with neurological impairment and granulocyte dysfunction. PYRIMIDINE 5’ NUCLEOTIDASE (P5’N-1) DEFICIENCY P5‘N-1, uridine monophosphate hydrolase-1 (UMPH-1) or cytosolic 5:nucleosidase II (cN-III) is an enzyme which major role is in the catabolism of the pyrimidine nucleotides, uridine monophosphate (UMP) and cytidine monophosphate (CMP), mainly resulting from RNA degradation during erythrocyte maturation. P5‘N-1 deficiency is an autosomal recessive disorder characterized by CHA with marked reticulocytosis and increased concentrations of pyrimidine nucleotides within mature erythrocytes. A characteristic RBC morphological abnormality is a heavy basophilic stippling, and its observation is very helpful for P5‘N diagnosis.P5‘N-1 deficiency can also be acquired as a result of lead poisoning or oxidative stress. Lead is a powerful inhibitor of P5‘N and determination of lead levels should be included whenever the constellation of haemolytic anaemia, P5‘N deficiency, and basophilic stippling is found. COMPILED BY Dr. V. MAGENDIRA MANI ASSISTANT PROFESSOR OF BIOCHEMISTRY ISLAMIAH COLLEGE (AUTONOMOUS) VANIYAMBADI, TAMILNADU, INDIA DOWNLOAD NOTES AT: http://tvuni.academia.edu/mvinayagam https://tvuni.academia.edu/mvinayagam Page14