South Korea medical device approval chart - Emergo
•
2 recomendaciones•4,941 vistas

Easy to understand chart describes the KFDA medical device registration process in South Korea.

Recomendados
Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Similar a South Korea medical device approval chart - Emergo
Similar a South Korea medical device approval chart - Emergo (20)
Último
Último (20)
South Korea medical device approval chart - Emergo
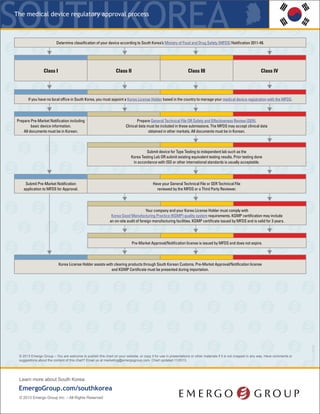
- 1. The regulatory process for medical devices KRSouth Korea * If your device (all classes) is new or does not have a predicate in the South Korean market, then the MFDS will require a Clinical Data Review, formerly Safety and Efficacy Review (SER). Manufacturers will be required to submit Clinical Data with their application and the registration approval timeline and fees will increase accordingly. ** There are approximately 350 Class II devices that MFDS considers “Special Equivalent,” which require a less robust application, i.e. no Technical File, as well as an expedited review by a Third Party Reviewer. *** Prior testing done in accordance with ISO or other international standards will satisfy some testing requirements; however, most manufacturers have to conduct additional testing, including performance testing, to Korea specific product standards. This is a simplified overview of the process. The MFDS may choose to audit your submission and request more documents, which will add time to your approval. © 2015 Emergo – You are welcome to publish this chart on your website, or copy it for use in presentations or other materials if it is not cropped in any way. Have comments or suggestions about the content of this chart? Email us at marketing@emergogroup.com. Chart updated 03/2015. 5040-0315 EmergoGroup.com/south-korea Determine classification of your device based on the device database, regulations provided by South Korea’s Ministry of Food and Drug Safety (MFDS), as well as predicate devices in the South Korean market. If no predicate exists, the device will follow the Clinical Data Review route.* Class I Class II** Class III Class IV Ifyou do not have a local office in South Korea,you must appoint a Korea License Holder based in the countryto manageyour medical device registration with the MFDS. Prepare Pre-Market Notification including basic device information. Prepare registration application in Korean, including technical file, test reports and KGMP certificate. Prepare registration application in Korean, including technical file, test reports and KGMP certificate. Class IV devices require an additional technical file in STED format. Send your device to Korea for testing OR prepare existing equivalent Korea testing results. *** Appoint a qualified importer/distributor to bring your device into South Korea. You are now authorized to begin marketing your device. KLH submits Pre-Market Notification application to the MFDS database. Pay application fees. KLHsubmitsKoreanregistrationdossierto acertifiedThirdPartyReviewer. Application includestechnicalfile,testreports,and KGMPCertificate. AftersuccessfulThird PartyReview,theMFDSwillissuethefinal approval.Payapplicationfees. KLH submits Korean registration dossier to the MFDS for review. Dossier includes the Technical File plus STED Technical File (Class IV), test reports, and KGMP certificate. Pay application fees. Obtain Korea Good Manufacturing Practices (KGMP) certificationthrough an onsite audit. Class II device manufacturing facilities are usuallyaudited byaThird PartyAuditor. KGMPcertification must be renewed every3years. Obtain Korea Good Manufacturing Practices (KGMP) certification through an onsite audit. Class III and IV device manufacturing facilities are audited by a Third Party Auditor and the MFDS. KGMPcertification must be renewed every3years. Class I devices do not require approval, only Notification to the MFDS. The MFDS will publish the device on their website. Notifications do not expire. Full application review conducted. Pre-Market Approval license is issued by the MFDS and published on their website. Registrations do not expire.
- 2. The regulatory process for medical devices EmergoGroup.com/south-korea This is a simplified overview of the process. South Korea’s Ministry of Food and Drug Safety (MFDS) may choose to audit your submission and request more documents, which will add time to your approval. NOTE 1: The time frames shown above are typical for the majority of medical device submissions but assume that your device does not contain animal tissue, medicinal substances or employ entirely novel technology. Your length of approval will depend on the quality and completeness of your technical documentation and how much time you take to address additional information requests from authorities after submission. YOUR SUBMISSION(S) MAY TAKE MORE TIME THAN WHAT IS SHOWN ABOVE. NOTE 2: The device license does not expire, but is subject to reevaluation. Periodically the MFDS will select devices codes to reevaluate as a whole. Typically this does not apply to products that were recently approved under that selected device code, as this process is intended to ensure older products meet the current regulatory requirements. Manufacturers will need to submit documents for reevaluation; documents include but are not limited to: updated vigilance, updated QMS documents, updated approvals from other markets. Announcements of device codes up for reevaluation typically happen in April or May, and manufacturers must submit before the end of the year. Failure to comply with the reevaluation will cause the device license to be revoked. NOTE 3: The device notification (Class I) and license (Class II-IV) does not expire, so no renewal is required. NOTE 4: Our rating of the complexity of the registration process is based on our experience and the opinion of nearly 1,000 QA/RA professionals worldwide who were asked to rate the difficulty of registering a device in each country. The European CE Marking process is considered the mid-point to which all other markets are compared. NOTE 5: Low = Less than US$5000; Midpoint = US$15000-$30000: High = More than US$50000. Overall cost includes registration application fees, product testing, in-country representation, submission preparation consulting and translation of registration documents but not IFU. Costs assume you already have approval for your device in the United States, Europe, Canada or Japan. Does not include cost of implementing or auditing a quality management system, if applicable. * If your device (all classes) is new or does not have a predicate in the South Korean market, then the MFDS will require a Clinical Data Review, formerly Safety and Efficacy Review (SER). Manufacturers will be required to submit Clinical Data with their application and the registration approval timeline and fees will increase accordingly. †† There are approximately 350 Class II devices that MFDS considers “Special Equivalent,” which require a less robust application, i.e. no Technical File, as well as an expedited review by a Third Party Reviewer. © 2015 Emergo – You are welcome to publish this chart on your website, or copy it for use in presentations or other materials if it is not cropped in any way. Have comments or suggestions about the content of this chart? Email us at marketing@emergogroup.com. Chart updated 03/2015. Device classification in South Korea How long you should expect to wait after submission until approval is granted. (See note 1) Validity period for device registrations. (See note 2) Registration renewal should be started this far in advance. (See note 3) Complexity of the registration process for this classification. (See note 4) Overall cost of gaining regulatory approval. (See note 5) CLASS I 1 month Does not expire Not applicable Simple Complex Low High CLASS II*† 4-6 months Does not expire Not applicable Simple Complex Low High CLASS III* 6-10 months Does not expire Not applicable Simple Complex Low High CLASS IV* 6-10 months Does not expire Not applicable Simple Complex Low High KRSouth Korea