2. Objectives
• Discuss a patient with A1AT deficiency
• Review genetics and pathophysiology of A1AT
deficiency
• Understand clinical symptoms, diagnosis,
monitoring, and treatment of A1AT deficiency
3. Case
• 8 month old boy diagnosed with A1AT on
screening because brother had A1AT
• Our patient presents with bloody emesis and
transfer to UCLA from OSH
• Hct drop from 33 to 27
• Patient also began to have melanotic stools
• Transferred after stabilization
4. Case
• PMH: BH unknown, no past hospitalizations or
surgeries
• SH: Lives with parents and brother
• Brother is 7 years old who presented with
neonatal hepatitis which resolved but found to be
ZZ for A1AT deficiency
• FH: negative for other types of liver disease
• NKDA
• Meds: none
5. Case
• Exam normal except for irritability and
“Abdomen: Distended with shifting dullness.
Spleen approximately 1cm to 2cm below the left
costal margin. Liver approximately 3 to 4cm
below right costal margin”
• Labs:
– CBC 10/9/25/140
– PTT 25.7 PT 10.9 INR 1.1 (after vitamin K)
– Chem 132/5.2/106
– Alb 2.6 ALT 143 AST 313 Tbili 0.7 Dbili 0.4
– Cr 0.1 Bun 10
6. Hospital Course
• 5/29/1997 to 6/10/1997
• Made NPO and put on octreotide
• EGD: small esophageal varices, sclerotherapy not
indicated, no active bleeding; snakeskin stomach
mucosa
• Started on nadolol, isosorbide, sucralfate, pepcid,
and aldactone
• MR venogram was normal
• US showed hyperechoic liver (likely cirrhotic),
splenomegaly, and ascites
• Discharged home on low Na and fluid restriction
7. Case
• No liver biopsy done but put on transplant list
• Received OLT on 10/24/1997
• Liver explant showed:
– bridging fibrosis
– Bile duct proliferation
– PAS stained globules / immunoperoxidase staining
for A1AT positive
8. Alpha 1-Antitrypsin
• Discovered in 1965 by Laurell and Eriksson
• Most common genetic cause of liver disease in
children
• Incidence of 1 in 2000-3000 in U.S.
• All races can be affected but most common in
whites
• Predisposes to chronic obstructive pulmonary
disease
• Serum levels of A1AT protein reduced
Maurice, Clin Transl Sci, 2012
9. Function of A1AT
• Serine protease inhibitor (SERPIN)
• Inhibits neutrophile elastase and other
protease
• Mainly synthesized in liver
10. Genetics
• Autosomal recessive
• Classic form is homozygous ZZ
• MM is the normal genotype (95% of population)
• S allele expressed 50-60% of protein
• Z allele expresses 10-20% of protein
• Z allele is common in northern Europeans
• MZ usually does not lead to liver disease
• More than 100 allelic variants
– Rare: Mmalton, Mduarte, null
PiSS
PiSZ
PiZZ
13. Liver Disease in A1AT
• Only 10% of homozygotes develop clinically
significant liver disease in first 3 decades
• Just over 1/3 of ZZ adults develop clinically
significant liver disease
• Progression of liver disease in variable
• No way to predict
• Abnormal protein (Z) accumulated in ER
• Can predispose to malignancy
Maurice, Clin Transl Sci, 2012
Perlmutter, Ann Rev Med, 2011
Fairbanks, Am J Gastro, 2008
17. Population-based studies indicate that 80% of PIZZ
patients presenting with neonatal cholestasis are healthy
and free of chronic disease by the age of 18 years.
Fairbanks, Am J Gastro, 2008
Sveger, Hepatology, 1995
Only 2-3% of ZZ
children progress
to needing liver
transplantation
during childhood.
18. Mechanism of Disease
• Mutant protein has altered
folding
• Tendency to polymerize and
aggregate
• Abnormal accumulation in
hepatocytes -> inflammation
-> fibrosis
• Other genetic and/or
environmental modifiers
– Impaired autophagy
– Infections? Viral hepatitis?
Alcohol?
Perlmutter, Hepatology, 2007
22. Associations
• Systemic vasculitis
• Interstitial fibrosis in patients with RA
• Relapsing panniculitis
• Multiple sclerosis
• Peripheral neuropathy
• Intracranial aneurysms
• ?IBD (studies weak)
• Glomerulonephritis
Fairbanks, Am J Gastro, 2008
23. Histology
• PAS (periodic acid Schiff)
stain accumulated protein
in ER
• Globule devoid
hepatocytes and
malignancy
• Nonspecific: giant cell
transformation, bile duct
paucity, bile duct
proliferation, lobular
hepatitis, steatosis,
fibrosis, necrosis
Fairbanks, Am J Gastro, 2008
24. Treatment
• No specific therapies for A1AT liver disease
• Cholestasis
– Fat soluble vitamins
– MCT oil
– Actigall
• Avoid smoking (including secondhand smoke) and
environmental pollution
• Monitor for HCC (AFP, ultrasound)
• OLT (also decreases or eliminates emphysema risk)
• Lung: IV purified pooled human plasma A1AT
(augmentation therapy)
26. Summary
• A1AT is a relatively common but under-
recognized and under-diagnosed genetic disease.
• Liver disease progression is quite variable and not
dependent on A1AT genotype alone.
• Liver disease mainly with ZZ and SZ (necessary
but not sufficient).
• No “cure” beyond OLT.
• Elucidation of pathophysiology of A1AT liver
disease is paving way to future therapies.
27. References
• Blanco, et al, “A1AT gene frequency distribution using maps of the world,” Hepatitis Monthly,
2012.
• Bornhorst, et al, “A1AT phenotypes and associated serum protein concentrations in a large
clinical population,” Chest, 2012.
• Clark, et al, “Liver test results do not identify liver disease in adults with A1AT,” Clin Gastro Hep,
2012.
• Fairbanks, “Liver disease in A1AT deficiency: a review,” Am J Gastro, 2008.
• Maurice and Perlmutter, “Novel treatment strategies for liver disease due to alpha 1-
antitrypsin deficiency,” Clin Transl Sci, Jun 2012.
• Nelson, et al, “Diagnosis and management of patients with A1AT,” Clin Gastroenterol Hepatol,
2012.
• Perlmutter, “A1AT deficiency: importance of proteasomal and autophagic degradative pathways
in disposal of liver disease-associated protein aggregates,” Annu Rev Med, 2011.
• Perlmutter, et al, “Molecular pathogenesis of A1AT deficiency associated liver disease,”
Hepatology 2007.
• Sveger and Eriksson, “The liver in adolescents with A1AT deficiency,” Hepatology, 1995.
• Sveger, “Liver disease in A1AT deficiency detected by screening 200,000 infants,” NEJM, 1976.
• Teckman and Lindblad, “A1AT deficiency: diagnosis, pathophysiology, and management,” Curr
Gastro Reports, 2006.
Editor's Notes
Asians and mexican american<black<hispanic<whiteClassic – northern european heritage
Alpha 1-antitrypsin (AAT) is the major serine protease inhibitor in human serum
Among whites, the most common allele, M, accounts for95% of alleles, while S and Z account for 2–3% and 1% of alleles,respectively. The most prevalent carrier phenotypes arePiMS and PiMZ, and deficiency phenotypes are PiSS, PiSZ,and PiZZ.Based on migration of the protein product in gel electrophoresis.Both Mmalton and Mduarte are associated with liver inclusions and clinically apparent liver disease. Null not associated with liver diease but lead to pulmonary manifestations.
A lot of adults will have normal LFTs!
A1AT concentration measured by nephelometryLook out for false negatives (acute phase reactant!) in heterozygous patients. ZZ usually low anyway in the face of inflammation.A1AT can be falsely low in substantial liver disease or malnutrition!
By adulthood most patients have normal liver enzymes and minimal or no sx of liver disease. At autopsy, however, 1/3 of ZZ patients have cirrhosis. (males>females)
By age 18, only 12 percent had elevations in serum alanine aminotransferase or gamma glutamyltransferase [5].
Autophagy = cellular mechanism for disposal of accumulated proteinRetained in ER endoplasmic reticulum of liver cellsThe downstream effects of the intracellular accumulation of the mutant Z protein include the formation of unique protein polymers, activation of autophagy, mitochondrial injury,endoplasmic reticulum stress, and caspase activation, which subsequently progress in a cascade, causing chronic hepatocellular injury.
Figure 2. The hypothetical hepatocellular injury pathway in PIZZ α1AT deficiency is depicted. The α1AT mutant Z protein is appropriately synthesized but then retained in the endoplasmic reticular (ER) of hepatocytes rather than being secreted. “Quality control” processes within the cells direct most of the mutant Z protein molecules into intracellular proteolysis pathways. However, some of the mutant Z protein molecules escape proteolysis and may attain a unique, polymerized conformation, forming inclusions in the ER. This results in activation of a variety of cellular responses. Autophagy maybe upregulated, resulting in damage to mitochondria; mitochondria may be damaged directly or may be involved in activation of caspases and the intrinsic pathway of apoptosis. Hepatocellular death may result from mitochondrial dysfunction or from an uninhibited apoptotic cascade. Given the variable nature of clinical liver injury between individuals with the same genotype, and the usually slow disease progression, there are likely to be important environmental and genetic disease modifiers affecting the rate and magnitude of hepatocellular death.
Necrotizing panniculitis – inflammatory lesions (typically older 40s)Hot, painful, red nodules or plaques on thigh or buttocks.Due to unopposed proteolysis in the skinSkin biopsy can show Z type AAT polymers in afffected areasTx: IV infusion of AAT, dapsone, doxycycline (scavenge reactive oxygen species)Vascular: unopposed proteolytic activity damages vascular wallsMembranoproliferative GN associated with ZZ (all pts usually have cirrhosis_ -> immune complex disorderIgA nephropathy which can be seen with hepatic cirrhosis regardless of etioogy (imapired removal of IgA complexes) by Kupffer cells of liver
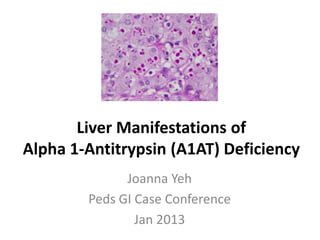
Figure 1. Liver biopsy photomicrographs from a 31-yr-old malewith PiZZ alpha 1-antitrypsin deficiency showing periodic acid-Schiff-positive diastase resistant globules (A) confirmed by immunoperoxidasestaining, magnification×128 (B) in hepatocyte cytoplasm,consistent with retained alpha 1-antitrypsin-Z molecules,magnification ×128.PAS globules can be absent in infants!
Ultrasound q6mo-1 year?
Figure 2. Effect of carbamazepine, fl uphenazine, and pimozide on intracellular degradation of mutant ATZ. Soluble formsof ATZ (purple) are degraded by the proteasomal pathway (top right) and insoluble aggregates of ATZ (gold) are degradedby autophagy (bottom right). Carbamazepine appears to enhance autophagy and also the proteasome. Fluphenazine andpimozide enhance the autophagic degradation of ATZsiRNA cloned into viral vectors and injected into portal vein