BLAST herramientas búsqueda similitud
•Descargar como DOCX, PDF•
0 recomendaciones•246 vistas


Recomendados
Más contenido relacionado
Similar a BLAST herramientas búsqueda similitud
Similar a BLAST herramientas búsqueda similitud (20)
BLAST herramientas búsqueda similitud
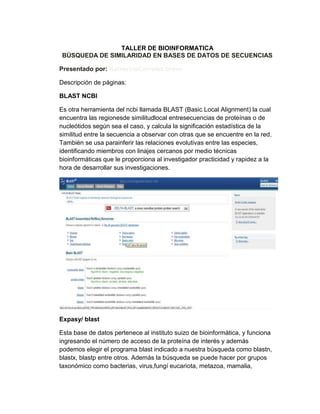
- 1. TALLER DE BIOINFORMATICA BÚSQUEDA DE SIMILARIDAD EN BASES DE DATOS DE SECUENCIAS Presentado por: KatherineCorrales bravo Descripción de páginas: BLAST NCBI Es otra herramienta del ncbi llamada BLAST (Basic Local Alignment) la cual encuentra las regionesde similitudlocal entresecuencias de proteínas o de nucleótidos según sea el caso, y calcula la significación estadística de la similitud entre la secuencia a observar con otras que se encuentre en la red. También se usa parainferir las relaciones evolutivas entre las especies, identificando miembros con linajes cercanos por medio técnicas bioinformáticas que le proporciona al investigador practicidad y rapidez a la hora de desarrollar sus investigaciones. Expasy/ blast Esta base de datos pertenece al instituto suizo de bioinformática, y funciona ingresando el número de acceso de la proteína de interés y además podemos elegir el programa blast indicado a nuestra búsqueda como blastn, blastx, blastp entre otros. Además la búsqueda se puede hacer por grupos taxonómico como bacterias, virus,fungí eucariota, metazoa, mamalia,
- 2. rodentia,Etc. que da estadísticas precisas en relación de similitud con la especie; la información se puede enviar por correo. Ha y la opción de una búsqueda avanzada, por categorías como las mejores alineaciones, alineación de huecos entre otros. Ch-EMBnet.org Ofrece al investigador la posibilidad de elegir entre blastn, blastx y blastp. Se pega la secuencia de proteínas que se quiera compara y esta la opción de dejar el correo electrónico para enviarle los resultados correspondientes y hay la opción de buscar en bases de datos de nucleótidos y proteínas.
- 3. NCBI BLAST El objetivo de esta herramienta es por medio de la comparación de secuencias encontrar regiones de similitud de secuencia que le indican al investigador pistas funcionales y evolutivas sobre la estrctura y función de una nueva secuencia. Se puede elegir varias bases de datos para mejorar la búsqueda como UniProtKnowledgebase, UniProtKB/Swiss-Prot, UniProtKB/Swiss-Protisoforms, UniProtKB/TrEMBL entre otras. Ab blast Por medio de una seria de algoritmos especiales, es uno de los más rápidos y actualizados buscadores sin requisitos especiales de software, o restricciones de uso. También tiene las opciones de BLASTP, BLASTN, BLASTX, TBLASTN, y TBLAST. Identifica los errores cuanto antes y ahorra tiempo.
- 4. Brassica BLAST server: El servicio se lleva a cabo por WU-BLAST2.0, según el parámetro que mejor convenga al investigador se escojeblastx, blastp y blastn, ademas de vaias bases de datos en las sé que pueden mejorar el sentido de la búsqueda. Se puede elegir el número de alineaciones, y el score respectivo de las secuencias. JCBI CMR Es una herramienta que permite al investigador u acceso a todas las secuencias del genoma bacteriano, o cualquier subconjunto de ellos. Al igual que el resto de herramientas biotecnológicas que hemos visto este también tiene muchas bases de datos en la cual puedo mejorar el sentido de mi búsqueda además de dar las alineaciones entre secuencias.
- 5. DDBJ Es una herramienta muy completa en la cual el investigador tiene la posibilidad de elegir entre el número de alineamiento que desea, el número de escores, si tiene filtro o no y demás detalles de la clasificación taxonómica del organismo a analizar. http://blast.ddbj.nig.ac.jp/blastn?lang=en 1) BLASTP EN EL NCBI Para poder analizar las proteínas descargamos el formato fasta que me brinda la secuencia de aminoácidos de la proteína a analizar.
- 6. En esta grafica vemos que tiene 4 proteínas que conserva mucha similitud con la proteína de nuestro interés mientas que ahí michas que se van alejando mas pero que de igual forma nos puede proporcionar información en un caso dado como la rojo, morado y verde. Son de relativa confiabilidad.
- 7. El listado amplia la información de la gráfica no dice los porcentajes exactos de la similitud entre secuencias, el nombre de las proteínas afines y además el valor de E value, que indica que entre menos valor más relacionada es la secuencia como se puede evidenciar son las secuencias de las líneas rojas que son casi idénticas, con la proteína de interés. Se concluye que nuestra secuencia es muy similar con Es similar a la proteína, el score que es la medida estadística del aproximado que se tiene en común ambas secuencias. De las proteínas C 23.
- 8. El blast nos muestra la estructura exacta de las secuenecias de aminoacidos mas similares con nuestra secuencia. Y ademas su grafica. Como podemos evidenciar la imagen de las dos secuencias son casi identicas
- 9. 2) BLASTP EN EMBnet Este buscador es muy similar al del ncbi con la direncia en que en el ncbi el resultado menos confiable era el negro aquí es el azul,y basicamente lo que tenemos en la grafica son 2 proteinas que tiene sus secuencias casi identicas con la de la proteina estudiada y son NUCL_MOUSE y NUCL_KAT.
- 10. Ademas me brinda la posibilidad de ver mas claramente los linajes cercanos con la ayuda de algoritmos que acomodan las proteinas en un mapa a modo de arbol que facilita el analisis deacuerdo a la similitud de estas. http://blast.ncbi.nlm.nih.gov/blast/treeview/treeView.cgi?request=page&blastR ID=RX1WCVS9015&queryID=lcl|62612&distmode=on&entrezLim=&ex=&exl= &exh=&ns=100&screenWidth=1280&screenHeight=1024
- 11. Con ayuda del blast de NCBI puedo comparar como varia la misma secuencia en ambos buscadores ya estas trabajan con distintos algoritmos .
- 12. En ambos buscadores las secuencias arrojan resultado idéntico. 3http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=tblastn&BLAST_PROGR AMS=tblastn&PAGE_TYPE=BlastSearch&SHOW_DEFAULTS=on&LINK_LO C=blasthome