Descargado 473 veces
























![8 Capítulo 1 La naturaleza de la fisícoquímíca y la teoría cinética de los gases
1.4 EQUILIBRIO TÉRMICO
Ley cero de la
termodinámica
Con frecuencia, al poner en contacto dos objetos de temperatura distinta durante
un periodo prolongado, sus temperaturas se igualan; alcanzan el equilibrio con
respecto a la temperatura. En este caso se utiliza el concepto de calor como forma
de energía. Se observa que el flujo de calor del cuerpo más caliente sirve para
aumentar la temperatura del cuerpo más frío. Sin embargo, el calor no es la
temperatura.
Se ampliará el concepto de equilibrio considerando dos cuerpos A y B que
están en equilibrio térmico entre sí, y al mismo tiempo un cuerpo adicional C que está
en equilibrio con B. Experimentalmente se ve que A y C también se encuentran en
equilibrio entre sí. Ésta es la ley cero de la termodinámica: dos cuerpos en equilibrio
térmico con un tercero también se encuentran en equilibrio entre sí. Esto permite
efectuar mediciones de temperatura.
El concepto de temperatura y su determinación
Escala Celsius
Las sensaciones físicas que se aceptan como indicaciones de que un objeto está
caliente o frío no sirven de manera cuantitativa: son de tipo relativo y cualitativo. El
primer termómetro que utilizó los puntos de congelación y de ebullición del agua
como referencia fue creado por el astrónomo danés Olaus Rerner (1644-1710). En la
vieja escala centígrada [del latín centum, cien; gradus, escalón; llamada también
escala Celsius en honor del astrónomo sueco Anders Celsius (1701-1744)], el punto
de congelación del agua a una atmósfera (atm) de presión se fijó a exactamente O°C
y el punto de ebullición a exactamente 100°C. Posteriormente se explicará por qué la
escala Celsius se define de manera algo distinta en la actualidad.
La construcción de diversos termómetros se basa en el hecho de que una
columna de mercurio cambia de longitud cuando se modifica su temperatura. En
algunos termómetros también se emplea la longitud de una varilla metálica sólida
o el volumen de un gas a presión constante. De hecho, para cualquier propiedad
termométrica ya sea que hubiera cambio de longitud o no, la vieja temperatura
centígrada e se relacionaba con dos temperaturas definidas. En el caso de una
columna de mercurio se asigna a su longitud el valor l¡oo cuando se encuentra en
equilibrio térmico con el vapor de agua en ebullición a una presión de 1 atm. El
punto en que se alcanza el equilibrio en hielo que se está fundiendo a una atmósfera
de presión sirve para establecer el valor de lo para esta longitud. Suponiendo que
haya una relación lineal entre la temperatura e y la propiedad tennométrica (la
longitud en este caso), y suponiendo que haya 100 divisiones entre las marcas fijas,
se puede escribir
(1.15)
donde 1 es la longitud a la temperatura e, y 10 Y 1¡00 son las longitudes a la
temperatura de congelación y de ebullición del agua, respectivamente. Algunas
propiedades termométricas no dependen de la longitud, por ejemplo los termóme-
tros de cuarzo que utilizan la frecuencia de resonancia del cristal de cuarzo como
propiedad termométrica. Sin embargo, también a ellos es aplicable la ecuación
1.15. Las propiedades termométricas de los materiales reales en general se desvían
de la linealidad exacta, inclusive en intevalos pequeños, debido a las interacciones
atómicas o moleculares dentro del material en sí, lo que reduce el valor de esa](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-25-320.jpg)





![14 Capítulo 1 La naturaleza de la fisicoquímica y la teor-ía cinética de los gases
En forma experimental, se observa que la ecuación 1.19 es válida para gases
reales a temperaturas de moderadas a altas sólo a medida que la presión tiende a cero.
Por lo tanto, la ley de Gay-Lussac, ecuación l.19, se podría expresar como
lím V= CT
P-.o
(l.20)
donde C es una constante. Esta expresión servirá como base para una nueva escala
de temperatura. Por lo tanto, si la temperatura y el volumen de una cantidad fija de
gas que se mantiene a cierta presión baja son TI y V], respectivamente, antes
de agregar calor, la relación entre la temperatura T2 y TI tras la adición de calor se
obtiene mediante la relación del volumen inicial y el volumen final, V2, del gas. En
consecuencia,
T2 = lím p --> oV2
TI lím p --> o VI
(l.21 )
Sin embargo, el límite de presión baja de un volumen de gas es infmito y, por lo tanto,
esta relación resulta poco práctica. En su lugar, se puede utilizar la siguiente expre-
sión:
T2 lím p --> o (PVh
TI límp--> o (PV)I
(1.22)
Gases ideales
Así, se tiene un termómetro de gas que trabajará igualmente bien con cualquier gas.
Si se supone que el gas obedece con exactitud la ecuación 1.19 o 1.22 para todos los
valores de P, entonces se ha definido un gas ideal y el termómetro que emplea un
gas de este tipo se denomina termómetro de gas ideal. Este concepto es de gran
utilidad para trabajar a bajas temperaturas.
Aunque se efectuaron esfuerzos considerables para definir una escala de tempe-
ratura con dos puntos de referencia como se indicó con anterioridad, el trabajo a baja
temperatura requiere de una escala que se base únicamente en un punto experimental
junto con el cero absoluto. En 1954 se tomó la decisión de redefinir la escala absoluta
y la de Celsius. El cero absoluto es el cero kelvin, representado como OK. Como las
medidas más cuidadosas del punto de congelación (el equilibrio entre agua y hielo a
una atmósfera de presión) varían en varios cientos de kelvins, entonces el punto triple
del agua (equilibrio entre agua y hielo y vapor de agua) se define como la temperatura
exacta de 273.16 K. Por lo tanto, el punto de congelación es casi exactamente
273.15 K y el punto de ebullición es tan solo otra temperatura que se mide en forma
experimental; equivale casi exactamente a 373.15 K o 100°C. El valor de la tempe-
ratura en kelvins se obtiene por definición sumando exactamente 273.15 al valor de
la temperatura en grados Celsius. Por lo que respecta a los intervalos de temperatura,
el grado Celsius es igual al kelvin.
Al emplear el valor definido para el punto triple como TI, una definición de
trabajo en la nueva escala Celsius es
Punto triple
lím p --> o (PV)T
T2=273.160
x . 2
lím p -. o (PV)puntotriple
(a P y n constantes) (l.23)
donde lím p -. o (PV)T = Ocuando T2 = O.](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-31-320.jpg)
![1.8 La ecuación de estado para un gas ideal 15
1.8 LA ECUACiÓN DE ESTADO PARA UN GAS IDEAL
La constante de los gases y el concepto de mol
En las secciones 1.5 y 1.6 se encontró que la presión, el volumen y la temperatura
están relacionados. Experimentalmente, estas tres propiedades de un gas no se pueden
elegir de manera arbitraria para describir el estado de una cantidad. determinada de
gas. (Los efectos de los campos gravitacional, eléctrico y magnético se ignoran en
este tratamiento.) Para una cantidad fija de gas, las propiedades fundamentales se
relacionan mediante una ecuación de estado. Sin embargo, experimentalmente uno
se encuentra que la relación lineal de la ley de Boyle (Ec. 1.16) para los datos P-V
sólo se alcanza a presiones muy bajas. Por lo tanto, en el límite de presión igual a
cero, todos los gases deben seguir la ley de Boyle con el mismo grado de precisión.
En consecuencia, es conveniente expresar la ley de Boyle como
lím (PV) = C'
P-40
(1.24)
Del mismo modo, la ley de Gay-Lussac puede expresarse en la forma de la ecuación
1.20. Al combinar ambas expresiones se obtiene
Hipótesis de Avogadro
lím (PV) = C" T
P-40
Para determinar el valor de la constante C ", se utiliza una importante hipótesis
propuesta en 1811 por el fisico italiano Amedeo Avogadro (1776-1856), la cual dice'
que un volumen dado de cualquier gas (a temperatura y presión constantes) debe
contener el mismo número de unidades independientes. Además, este investigador
especificó que las partículas del gas pueden ser átomos o combinaciones de átomos,
y propuso la palabra molécula para estas últimas. La hipótesis de Avogadro dice que
(1.25)
I Vocn o V=C"'n (válida a P y T constantes) I (1.26)
Definición de mol
donde n es la cantidad de sustancia, que en el sistema SI se expresa en moles.
Un mol es la cantidad de cualquier sustancia que contiene el mismo número de
entidades elementales (átomos, moléculas, iones, etc.) que el que hay en exactamente
0.012 kg de carbono 12 (véase también Apéndice A). El número de entidades
elementales se relaciona con la cantidad de sustancia mediante la constante de
Avogadro; ésta se representa con el símbolo L [por Joseph Loschmidt (1821-1895),
quien fue el primero en medir su magnitud] y tiene el valor de 6.022 137 x l 023
mol ".
El valor numérico de la constante de Avogadro, 6.022 137 x 1023
, es el número de
entidades elementales en un mol.
Como las ecuaciones 1.20, 1.24 Y1.26 indican que el volumen de un gas depende
de T, l/P y n, respectivamente, estas tres expresiones pueden combinarse para dar
lím (PV) = n RT
P-40
(1.27)
Si se toman en cuenta las limitaciones que impone el requisito del límite, -4 o, la
ecuación puede escribirse de manera aproximada como
Ecuación de estado
de un gas ideal
(1.28)](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-32-320.jpg)


![número de colisiones en la unidad de tiempo = u,
2x
(1.32)
18 Capítulo 1 La naturaleza de la fisicoquímica y la teoría cinética de los gases
La fuerza Fes igual al cambio de momento p de la molécula en una dirección dada
por unidad de tiempo, de acuerdo con la segunda ley de Newton del movimiento,
dp d(mu) du
F=-=ma=--=m-
dt dt dt
(1.30)
El momento de la molécula en el sentido del eje X cuando choca contra la pared es
mu.. Si se supone que la colisión es perfectamente elástica, la molécula rebotará de
la pared a la velocidad -Ux en sentido opuesto.
El cambio de velocidad de la molécula en cada colisión es
/).11., = [-u, (después de la colisión)] - [ux (antes de la colisión)]
= -211., (1.31)
El cambio de momento correspondiente es -2mux' Ya que cada colisión con la pared
se produce sólo después de que la molécula, se desplaza una distancia 2x (es decir,
efectúa un viaje redondo), el número de colisiones que realiza la molécula por unidad
de tiempo puede calcularse dividiendo la distancia u, que la molécula viaja en la
unidad de tiempo, por 2x. El resultado es
El cambio de momento por unidad de tiempo es. por tanto,
o
du [11,) mu;F = 11/ - = (-2u,m) -'-- = --'
dt 2x x
(1.33)
La.fuerza F" que ejerce la partícula sobre la pared es exactamente igual en magnitud
a este valor, pero con signo opuesto:
?
I17U~
F".=-F=-
x
(1.34)
Como la presión es la fuerza por unidad de área y el área A es j.z, la presión en el
sentido del eje X, P" se puede expresar como
p = F'I' = FIr
X A 1'Z
o o
I17U~ I17U~
xy.:: l'
(1.35)
porque Xl'.:: = Ves el volumen del recipiente.
La presión de un gas derivada de la teoría cinética
Hasta el momento sólo se ha estudiado una molécula que supuestamente se desplaza
a velocidad constante.Un conjunto de :Vmoléculas tendrá una distribución de velo-](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-35-320.jpg)







![26 CapítlJlo 1 La naturaleza de la fisicoquímica y la teoría cinética de los gases
Trayectoria libre media
.l!
A 3000 K, ZN
2
, O
2
= 8.49 x 1034
m-3 S-l. A.partir de este ejemplo se observa
que el efecto de T sobre Z no es grande, ya que, según la ecuación 1.43 y el
último análisis, T aparece como rr.El efecto de d es mucho más profundo ya
que aparece como d2
•
»,
. "
Un concepto de particular importancia al considerar ciertas propiedades es la
trayectoria libre media A. Ésta es la distancia promedio que se desplaza una
molécula entre dos colisiones sucesivas. Se ha visto que para un solo gas, el número
de colisiones que efectúa una molécula por unidad de tiempo ZA, es {27rd;uANAIV
(Ec. 1.66). En el tiempo unitario la molécula viaja en promedio la distancia UA'
Entonces la trayectoria libre media es
distancia viajada por unidad de tiempo
A=------~~~----------~
número de colisiones en ese tiempo
v
La magnitud de los valores de dA evidentemente es de gran importancia con
respecto a la teoría cinética de los gases, ya que es la única propiedad molecular que
se necesita conocer para calcular los números de colisión y la trayectoria libre media .
.EJEMPLO 1.4 El oxígeno moleoular tiene un diámetro de colisión. de 3.57
por 10-10 m. Calcule A para el oxígeno a 300 K ya 101.325 kPa.
Solución Como PV = nRT = (N/L)RT, A puede expresarse como
RT
A=----
{27rd2LP
8.314 (1 K-1
mol'") x ~OO(:{<.)
=--------------------------------------------~
--J27r [3.57 x 1(JIO (m)]2 x 6.022 x 1023 (mol'") x 10 1325 (Pa)
= 7.22 X 10-8
m
ya que lIPa = m3
(véase Ap. A).
1.10 LA LEY DE LA DISTRIBUCiÓN BAROMÉTRICA
Se ha mencionado que se puede producir un cambio en las propiedades del sistema
aplicando un campo potencial a él. En esta sección se considerará el efecto de un
campo gravitacional. En los experimentos de laboratorio, los efectos de los campos
gravitacionales en general son ignoradas. Sin embargo, en sistemas a gran escala,
como la atmósfera terrestre o un océano, la gravedad ocasiona una variación notable
de propiedades Un ejemplo es el gran aumento de presión hidrostática en regiones
profundas del océano.
(1.67)
(l.68)](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-43-320.jpg)



![1.1
1.0
0.9
0.8
M
0.7
I
E
0.6()
en
a 0.5
"O
<1l
"O 0.4.¡¡;
e
~
al
0.3O
0.2
80atm
0.1
40 80 120 160
Temperatura, TrC
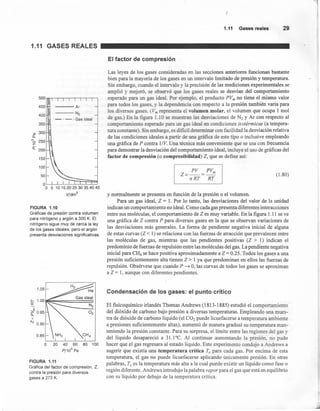
FIGURA 1.14
Variación de la densidad del C02
con la temperatura y la presión
total. La presión atmosférica total
que se menciona es la presión
calibrada. [Datos tomados de J.J.
Langenfeld et al., Anal. Chem. 64,
2265 (1992)]
1.11 Gases reales 31
TABLA 1.3 Constantes críticas para algunos gases
Te Pe Ve
Sustancia K bar dm3
mol"
H2 33.2 12.97 0.0650
He 5.3 2.29 0.0577
N2 126.0 33.9 0.0900
O2 154.3 50.4 0.0744
Ch 417 77.1 0.123
Ar 151 48.6 0.0752
Kr 210.6 54.9 0.092
CO 134 35.5 0.040
NO 183 65.9 0.058
CO2 304.16 73.9 0.0956
HCl 325 82.7 0.0862
S02 430 78.7 0.123
H20 647.1 220.6 0.0450
NH3 405.5 113.0 0.0723
CH4 190.6 46.4 0.0988
CChF2 385.1 41.1 0.217
CSH'2 469.8 33.7
C6H'4 507.4 30.3
contenido en un recipiente cerrado en la condición que se representa con el punto D
de la figura 1.12; la temperatura es T3' Ahora el líquido se calienta por encima de la
temperatura crítica hasta el punto F. Se mantiene el recipiente fijo en T5 mediante un
termostato y se permite que el volumen aumente hasta E. No se observa cambio de
fase durante estos procesos. Después, se deja que la temperatura descienda hasta la
isoterma T3 en el punto A. De nuevo no se observa cambio de fase. Ahora el sistema
se encuentra en estado gaseoso sin haber experimentado un cambio discontinuo de
fase. Por lo tanto, hay una continuidad total de estados en la cual, la transformación
de gas a líquido ocurre continuamente. En vista de lo anterior, la diferencia entre los
estados líquido y gaseoso sólo puede determinarse cuando coexisten dos fases.
Aplicaciones de los fluidos supercríticos
Los gases y líquidos en la región supercrítica se denominanfluidos supercriticos. Se
forman calentando una sustancia por arriba de su temperatura crítica, y tienen
propiedades significativamente distintas de las que presentan en estado normal. Por
ejemplo, las densidades de los gases en forma de fluidos supercríticos pueden
aumentar a más de 1.0 cm-3. Obsérvese la figura 1.14, en la cual se grafica la varíación
de la densidad del CO2 contra la temperatura a diversas presiones. Esta alta densidad
está asociada con la capacidad de algunos fluidos supercríticos para disolver molé-
culas no volátiles de gran tamaño. Esto permite el desarrollo de aplicaciones indus-
triales muy interesantes, como la eliminación de la cafeina de los granos de café
mediante dióxido de carbono supercrítico (evitando así el uso de hidrocarburos
cIorados, que son dañinos para el medio ambiente).](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-48-320.jpg)




![Ecuación de Berthelot
Ecuación de Dieterici
Ecuación de Redlich y
Kwong
1.13 La ecuación de virial 37
Otras ecuaciones de estado
Hay otras dos importantes ecuaciones de estado de uso común. P.A. Daniel Berthelot
(1865-1927) desarrolló la expresión
(1.94)
que es la ecuación de van der Waals modificada para la dependencia del término de
atracción con respecto a la temperatura. Puede expresarse de manera ligeramente
modificada en relación con las variables reducidas como:
p_(RTJ[1 +_9 -~pJ
-lVm 128Tr 64T; r
(1.95)
10que permite una precisión más alta a bajas presiones y temperaturas.
Otra ecuación de estado importante es la introducida en 1899 por C. Dieterici.
La ecuación de Dieterici incluye el número trascendental e (base de los logaritmos
naturales) y, por 10 tanto, es menos fácil de usar que la ecuación anterior; sin embargo,
brinda una mejor representación que las otras expresiones cerca del punto critico.
Puede expresarse como
(1.96)
donde a y b son constantes no necesariamente iguales a las de van der Waals. En
forma reducida, la ecuación 1.96 se transforma en
t; ( 2 JPr=---exp 2---
2Vr- 1 rv,
(1.97)
Se han propuesto otras ecuaciones. En 1949 Otto Redlich y Joseph N.S. Kwong
introdujeron la ecuación
[
p + 1/2 rt-a ] (V - bn) = nRT
T V(V+nb)
(1.98)
que es una expresión sencilla y precisa de dos parámetros, aplicable a una amplia
gama de temperaturas y presiones.
Otra expresión que ha ganado popularidad es la ecuación de estado de Benedict-
Webb-Rubin, la cual relaciona la presión con la densidad molar y la temperatura. Se
ha utilizado para predecir con bastante precisión las propiedades termodinámicas
de hidrocarburos complejos.
1.13 LA ECUACiÓN DE VIRIAL
La ventaja de las ecuaciones descritas en las últimas secciones es que las constantes
se mantienen al mínimo y se relacionan con parámetros defmidos en teoría. Otra](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-54-320.jpg)

![1.13 La ecuación de virial 39
EJEMPLO 1.5 Evalúe la temperatura de Boyle en términos de las constantes
conocidas A. by R para un gas que tiene Ia ecuación de estado
Solución La temperatura de Boyle se presenta cuando el segundo coeficiente
de virial, B(T) = O. Esta condición se cumple cuando la cantidad ib - AlRT2I3)
= O. En esta condición, T = TB• Despejando TB
Tf3=A/bR; TB = (A/bR)312
- ,
La importancia de los coeficientes de virial reside en el hecho de que, a través
de los métodos de mecánica estadística, la ecuación de estado de un gas real se puede
desarrollar en forma de virial. Los coeficientes derivados empíricamente se relacio-
nan así con sus contrapartes teóricas, que son (en último término) energías potenciales
intermoleculares. En esta interpretación los segundos coeficientes de virial, por
ejemplo, se deben a las interacciones entre pares de moléculas; los otros coeficientes
se deben a interacciones de orden más alto.
La ecuación de virial no es de particular utilidad a altas presiones o cerca del
punto crítico, porque la serie de potencias no converge con rapidez en condiciones
de interacciones de orden más alto. Asimismo, es más dificil proceder basándose en
la teoría que en forma empírica, y el cálculo de las constantes a partir de la
mecánica estadística se dificulta porque no se conocen bien las funciones potenciales
y la evaluación de las integrales múltiples que aparecen es muy dificil.
La expresión que se considerará en último término es la ecuación propuesta en
1927-1928 por los químicos estadounidenses James Alexander Beattie y Oscar C.
Bridgeman.i
Ecuación de
Beattie-Bridgeman [
p~r [1 - (c/v,,,r3
)] A
p= (Vm+B)--
v,; V~
(1.1 02)
donde
l
a
A=Ao 1--)
V",
B=BO(I-~J
l Vnl
y a, b. Aa. Bo Y e son constantes que se determinan empíricamente. La ecuación de
Beartie-Bridgeman utiliza cinco constantes además de R y es conveniente para
trabajos precisos, en especial en el intervalo de altas presiones. En la tabla 1.5 se dan
las constantes de Beattie-Bridgeman" para 10 gases.
~ lA Beattie y OC Bridgernan, J Am. Chem. Soc. . ./9, 1665 (1927): 50, 3133, 3151 (1928).
,¡ .1.- Beanie O e Bridgeman. Proc. Am. Acad. Arts Sci., 63,229 (1928).](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-56-320.jpg)



![1.27. Exactamente 1 dnr' de nitrógeno a presión de 1 bar, tarda
5.80 minutos en experimentar efusión a través de un orificio.
¿Cuánto tardará el He para experimentar efusión en las mismas
condiciones?
1.28. ¿Cuál es la energía cinética total de 0.50 mol de un gas
mono atómico ideal confinado a 8.0 dnr' a 200 kPa?
1.29. Se mantiene nitrógeno gaseoso a 152 kPa en un recipiente
de 2.00 dnr' a 298.15 K. Si su masa molar es 28.0134 g mol "
calcule:
a) La cantidad de N2 presente.
b) El número de moléculas presentes.
e) La media de la raíz cuadrada de la velocidad de las moléculas.
d) La energía cinética traslacional promedio de cada molécula.
e) La energía cinética traslacional total del sistema.
1.30. ¿Por qué factor se modifica la media de la raíz cuadrada
de las velocidades cuando un gas se calienta de 300 K a 400 K?
*1.31. El diámetro de colisión del Nz es 3.74 x 10-10 m a
298.15 K Y 101.325 kPa. Su velocidad promedio es 474.6 m S-l.
Calcule la trayectoria libre media, el número promedio de coli-
siones ZA que experimenta una molécula en la unidad de tiempo,
y el número promedio de colisiones ZAA por volumen unitario
por unidad de tiempo para el Nz.
*1.32. Exprese la trayectoria libre media de un gas en términos
de las variables presión y temperatura, que se miden con más
facilidad que el volumen.
1.33. Calcule ZA y ZAA para el argón a 25°C y una presión de
1.00 bar empleando el valor d = 3.84 X 10-10 m obtenido por
mediciones de cristalografia de rayos X.
1.34. Calcule la trayectoria libre media del AI a 20°C y 1.00 bar.
El diámetro de colisión es d = 3.84 X 10-10 m.
1.35. El hidrógeno gaseoso tiene un diámetro de colisión mo-
lecular de 0.258 nm. Calcule la trayectoria libre media del
hidrógeno a 298.15 K Y a) 133.32 Pa, b) 101.325 kPa y c) 1.0
X 108
Pa.
1.36. En el espacio interestelar se estima que existe hidrógeno
atómico a la concentración de una partícula por metro cúbico.
Si el diámetro de colisión es 2.5 x 10-10m, calcule la trayectoria
libre media A. La temperatura del espacio interestelar es 2.7 K.
*1.37. Se desea calcular el valor de la constante de Avogadro a
partir de un estudio efectuado por Perrin [Ann. Chim. Phys., 18,
1 (1909)] en el cual se midió, en función de la altura, la distri-
bución de las partículas coloidal es de color amarillo brillante
de una resina de goma en suspensión acuosa. Algunos datos a
15°C son
altura, z/l 0--<>
N, número relativo de partículas
de resina de goma a la altura z
Presina de goma = 1.206 g cm-
3
5 35
100 47
Problemas 43
Pagua = 0.999 g cm'?
radio de las partículas de resina de goma, r = 0.212 x 10--<>m.
(Sugerencia: Considere que las partículas son moléculas
gaseosas en una columna de aire y que el número de partículas
es proporcional a la presión.)
Gases reales
1.38. Dibuje la isoterma PV de van der Waals en el rnísmo
intervalo de P y V que en la figura 1.10 a 350 K Y 450 K para
CI2 utilizando los valores de la tabla 1.3.
, 1.39. Compare las presiones que se pronostican para 0.8 dnr' de
CIz que pesa 17.5 g a 273.15 K empleando a) la ecuación de los
gases ideales y b) la ecuación de van der Waals.
1.40. El etileno (C2lL¡) tiene una presión crítica de P, = 61.659
atm y una temperatura crítica de Te = 308.6 K. Calcule el
volumen molar del gas a T= 97.2°C y 90.0 atm empleando la
figura 1.16. Compare el valor encontrado con el calculado a
partir de la ecuación de los gases ideales.
1.41. Determine la temperatura de Boyle en términos de las
constantes para la ecuación de estado
PVm = RT¡l + 8/57 (PIPJ(TjT) [1- 4 (TjT)2]1
R, r.Y Te son constantes.
*1.42. Determine la temperatura de Boyle de un gas de van der
Waals en términos de las constantes a, b y R.
1.43. La temperatura crítica Te del óxido nitroso CN20) es de
36.5°C y su presión crítica P, de 71.7 atm. Suponga que 1 mol
de N20 se comprime a 54.0 atrn y 356 K. Calcule la temperatura
y la presión reducidas y utilice la figura 1.16, interpolando en
caso necesario, para estimar el volumen que ocupa 1 mol del gas
a 54.0 atrn y 356 K.
*1.44. Para la ecuación de Dieterici, derive la relación entre a
y b y el volumen y la temperatura críticos. [Sugerencia.' Recuer-
de que en el punto crítico (dP/dV)T= OY(d2PldV2h = O.]
*1.45. Un requisito general de todas las ecuaciones de estado
para gases es que se reducen a la ecuación de los gases ideales
(Ec. 1.26) en el límite de presiones bajas. Demuestre que esto es
cierto para la ecuación de van der Waals.
1.46. Las constantes de van der Waals para C2H6 en la literatura
antigua son
a = 5.49 atm emoJ-2
y b = 0.0638 L mol "
Exprese estas constantes en unidades SI (L = litro = drrr').](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-60-320.jpg)








![,...
52 Capítulo2 La primeraley de la termodinámica
FIGURA 2.2
El trabajo reversible efectuado por una
presión constante P que desplaza un
pistón. Una manera sencilla de que un
gas se encuentre a presión constante,
es tener un vapor en equilibrio con su
líquido,
Trabajo reversible que se efectúa
sobre el sistema w,ev = PAI = - P tiV
,.I
I
I
,
,,
presión I
constante P I
I
'
Disminución
1 I de volumen,
I :-_-_-_-_-_-_-_-_- __ -_~
,1 , -t1V= Al
,. ,
I
I
/ I
/
Presión
- aplicada
~------' = P+ dP
Gasa
Área de sección transversal = A
Wrev == F 1== P Al (2.8)
Sin embargo, Al es el volumen redonda que barre el desplazamiento del pistón, es
decir, la disminución del volumen del gas, que es - ~v.Por lo tanto, el trabajo que
se realiza sobre el sistema es
(2.9)
En el ejemplo ésta es una cantidad positiva, ya que se comprime el gas y ~V es
negativo. Si el gas se hubiera expandido, ~V sería positivo y el trabajo efectuado
sobre el sistema, negativo; es decir, el gas habría realizado una cantidad positiva de
trabajo sobre los alrededores.
EJEMPLO 1. Suponga que ocurre una reacción química en un bulbo unido
a un tubo capilar que tiene un área de sección transversal de 2.50 mnr', El tubo
está abierto a la atmósfera (presión = 101.325 kPa),y en el curso de la reacción
el volumen del capilar aumenta 2.4 cm. Calcule el trabajo efectuado' por el
sistema de reacción.
Solución El aumento de volumen es
2.50 X 10--6m2 x 2.40 x 10-2 m == 6.00 x 10-8 m3
El trabajo realizado por el sistema, que según la convención de la IUPAC debe
expresarse como -w, es P ~V:
-w = P áV= 1.01325 x lOS Pa x 6.00 x 10- 8 m3
= 6.08 x 10-3
J [Pa =N m-2; N m = 1]
Cuando la presión P varia durante el cambio de volumen, es necesario determinar ¡'
mediante integración el trabajo efectuado. El trabajo realizado sobre el sistema](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-69-320.jpg)










![66 CapíbJlo2 La primera ley de la termodinámica
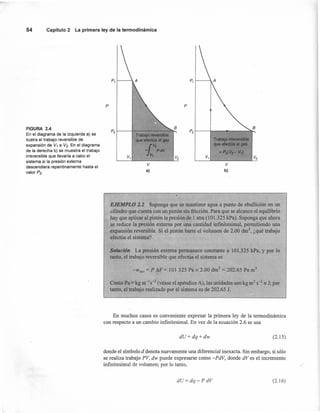
TABLA 2.1 Parámetros para la ecuación Cp,m = d + eT + rr«
Sustancia Estado d e f
J K-' mor' J K-
2
mor' J K mor'
He, Ne, AI, Kr, Xe Gas 20.79 O O
H2· Gas 27.28 3.26 x 10-3
5.0 X 104
.
O2 Gas 29.96 4.18 x 10-3
-1.67 X 105
N2 Gas 28.58 3.76 x 10-3
-5.0 X 104
CO Gas 28.41 4.10 x 10-3
-4.6 X 104
CO2 Gas 44.22 8.79 x 10-3 -8.62 X 105
H20 Vapor 30.54 10.29 x 10-3
O
H20 Líquido 75.48 O O
C (grafito) Sólido 16.86 4.77 x 10-3
-8.54 X 105
NaCl Sólido 45.94 16.32 x 10-3
O
Cuando se conoce á H(TI) para TI = 25°C, se puede obtener á Hm(T2) a cualquier
temperatura T2 mediante esta ecuación, y sustituyendo la ecuación 2.49 se tiene
(2.51)
_ 1 2 2 [1 lJ- !::'Hm(TI) +!::. d(T2 - TI) + - !::.e(T2 - TI) -!::.f ---
2 T2 TI
(2.52)
EJEMPLO 2.6 Considere la reacción en fase gaseosa
2CO(g) + 02(g) ~ 2C02(g)
Un estudio de esta reacción en una bomba calorimétrica a 25°C da los valores
de á H ° = -565.98 kJ mol ". Calcule A H ° para esta reacción a 2000 K.
Solución A partir de los valores de la tabla 2.1 se obtiene
.!::.d= d(productos) - d(reactivos)
= (2 x 44.22) - (2 x 28.41) - 29.96 = 1.66 J K-I
mol-I
!::.e = e(producto s) - e(reactivos)
= (2 x 8.79 x 10-3
) -(2 x 4.10 x 10-3
) -4.18 X 10-3
. = 5.29 x 10-3 J K-2 mol-I.
!::.f =f(productos) - j(reactivos)
= [2 x (-8.62 x 105)] + (2 x 0.46 x 105) + 1.67 X 105
= -14.65 X 105
J K mol'"
., ,](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-81-320.jpg)





![2.6 Relacionesde los gasesideales 73
(2.59)
El cambio de energía interna molar !:J.Um (también negativa para este proceso) se
obtiene aplicando la primera ley:
!:J.Um= q + w = ep• m (T2 - TI) + R(TI - T2)
= (ep' m - R)(T2 - TI) = eV, m (T2 - TI)
(2.60)
(2.61)
mediante la ecuación 2.36. Se puede verificar fácilmente que estas expresiones para
¡}J{m Y!:J.Umson congruentes con la relación
(2.62) '+,
Cambio de presión reversible a volumen constante
Supóngase, por el contrario, que 1 mol de gas ideal se lleva del estado inicial PI, Vm.¡,
TI al estado fmal P2, Vm.¡, T2 como se muestra en la figura 2.6b). La presión P¡ es
superior a P2 y para efectuar esto a volumen constante es necesario eliminar calor
hasta la temperatura T2• De nuevo, el cambio se efectúa de manera reversible.
El trabajo que se lleva a cabo sobre el sistema es el área debajo de la línea Ae
de la figura 2.6b) Y es cero. Esto se confirma por medio de la integral
r-W =- PdV=Orey V
m.1
(2.63)
Como el proceso ocurre a volumen constante, el calor absorbido es
(2.64)
que es negativo porque TI > T2• Esta expresión también es !:J.Um:
(2.65)
Se puede verificar que las ecuaciones 2.63, 2.64 Y 2.65 son congruentes con la primera
ley. El valor de !:J.Hm se obtiene como sigue:
!:J.Hm = !:J.Um+!:J. (PVm) = !:J.Um+!:J. (RT)
= eV, m (T2 - TI) + R(T2 - TI) = (ev, m + R)(T2 - TI)
I!:J. Hm = ep. m (T2 - TI)I
(2.66)
(2.67)
(2.68)
Es interesante comparar los cambios que se producen al pasar de A a B [dismi-
nución de volumen a presión constante, Fig. 2.6a)] con los que se producen al ir de
A a e [disminución de presión a volumen constante, Fig. 2.6b)]. El trabajo y los
valores calorificos son diferentes en ambos casos. Sin embargo, los valores !:J. Um y
!:J.Hm son iguales. Esto significa que la energía interna es la misma en el punto B sobre
la isotenna T2 que en el punto e; lo mismo ocurre con la entalpía. Este resultado se](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-88-320.jpg)




![1. PV=nRT
78 Capítulo2 La primeraley de la termodinámica
En 1845 Joule publicó los resultados de un experimento diseñado para determinar si
Tapón poroso", ( aUI aV)T = O para un gas real. Su aparato, que se muestra en la figura 2.7a), tenía
Estado3 P1 ~----- dos recipientes conectados a través de una llave y estaba colocado dentro de un baño
inicial V, Tt
. {----- de agua cuya temperatura se podía medir con gran precisión. Llenó un recipiente con
----,~ ~ aire a presión de 22 bares y evacuó el otro. Al abrir la llave de paso que conectaba am-
Estado Pz T.
2
final .V
2
. bos recipientes, no pudo detectar cambio alguno en la temperatura.
'----=---==-- La expansión en el experimento de Joule fue un proceso irreversible. El gas no
b)
experimentó cambio detectable de energía interna porque no se efectuó trabajo ni
FIGURA 2.7 hubo intercambio detectable de calor con los alrededores. El cambio dU puede
a) Aparato de Joule para medir un expresarse como (véase apéndice C)
cambio de temperatura al efectuarse la
expansión libre de un gas. b)
Experimento de Joule-Thomson: el gas
se hace pasar a través de una
membrana y se mide el cambio de
temperatura.
~R··,• I 11 "'
Airea
·22atm Vacío
a)
(2.98)
(2.99)
La segunda de estas condiciones se deduce de la primera. Un gas no ideal no cumple
ninguna de estas condiciones.
El experimento de Joule- Thomson
(2.1 00)
Joule llegó a la conclusión de que dU = O y, en consecuencia, que
(~~l= - (~~l(~~l
=-cv(~~l
(2.1 01)
(2.102)
según la ecuación 2.26. El experimento de Joule no indicó ningún cambio de
temperatura [o sea, (a TI aV)u = O]y, por lo tanto,
(auJ- -O
av T
(2.103)
En este experimento, Joule detectó que el gas se comportaba de manera ideal.
Sin embargo, dicho experimento no resultó muy satisfactorio porque la capaci-
dad calorífica del agua es sumamente grande en comparación con la capacidad
calorífica del gas, de manera que cualquier aumento de temperatura sería muy
pequeño y dificil de detectar. William Thomson (lord Kelvin) sugirió un mejor
procedimiento y, junto con Joule, efectuó una serie de experimentos entre 1852 y
1862. Su aparato, que se muestra de manera diagramática en la figura 2.7b), consta
fundamentalmente de un cilindro que tiene untapón poroso. Mediante un pistón,el
gas que se encontraba de un lado del tapón se hizo pasar por la fuerza a través de éste
a una presión constante p], y el segundo pistón se desplazó hacia atrás manteniendo](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-93-320.jpg)

![80 Capítulo2 La primeraley de la termodinámica
(2.111)
Una ecuación para la diferencia entre las capacidades calorificas Cp y C»
aplicable a gases, líquidos y sólidos, se obtiene como sigue:
(2.112)
También se obtienen las siguientes relaciones a partir de las diferenciales totales de
Uy V:
dU =(au] dV + (au] dT
av T aT v
(2.113)
y
dV= (aV] dT+ (av] dP
aT p ap T
Sustituyendo esta expresión para dVen la ecuación 2.113 se obtiene
(2.114)
, ¡ ¡~
dU=(au] (av] .dT+(au] (~V] dP+(au] dT
av T aT p av T ap T aT v
A presión constante se puede eliminar el segundo término del lado derecho:
(2.115)
(2.116)
Sustituyendo esta expresión en la ecuación 2.112 se obtiene
(2.117)
Para un gas ideal, (aUIdVh = Oy, por tanto,
Cp - Cv= P (av)
aT p
(2.118)
Presión interna
Para 1 mol de un gas ideal, PVm= RTy, en consecuencia (aVm/aT)p = RlP; así,
Cp.m-Cv.m=R (2.119)
como se probó con anterioridad en la ecuación 2.36.
En general, el término p(aV/aT)p de la ecuación 2.117 representa la contribución
a Cp que se debe al cambio de volumen del sistema que actúa en contra de una presión
externa P El otro término (aUlaV)T (av/aT)p, es una contribución adicional debida
a (aUlaV)n que es una presión efectiva surgida de las fuerzas de atracción o repulsión
entre las moléculas; la cantidad (aUlaV)r se denomina 'presión interna. En un gas
ideal la presión interna vale cero, y en los gases reales el término suele ser pequeño
en comparación con la presión externa P. Por otra parte, los líquidos y sólidos tienen
fuerzas de cohesión o de atracción muy altas y la presión interna (aUIdV)T será, por
tanto, grande en comparación con la presión externa.](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-95-320.jpg)
= nRT o (P+ ~l(Vm-b)=RT (2.120)
donde Vm. que es igual a Vln, es el volumen molar. Resulta evidente que las dos
relaciones válidas para un gas ideal, ecuaciones 2.98 y 2.99, no son válidas para un
gas de van der Waals. En lugar de la ecuación 2.98 se tiene la ecuación 2.120, y en
vez de la ecuación 2.99 se puede aplicar la ecuación
(~~l= n:~= :;
(2.121)
Esta relación se deriva en la sección 3.8 (pp. 128-129).
EJEMPLO 2.10 Suponga que un gas sigue la ecuación modificada de van-
derWaals
P(Vm -b) = RT
Y que b tiene el valor de 0.02 dnr' mol ". Si se comprimen 0.50 moles del gas
de manera reversible desde un volumen inicial de 2.0 dm3 hasta un volumen
final de 0.50 dnr', ¿cuánto trabajo se realiza sobre el sistema? ¿Cuánto trabajo
se habría realizado si el gas fuera ideal? Explique la diferencia entre los dos
valores.
Solución La ecuación aplicable para n moles de gas es
P(V - nb) = nRT
Según la ecuación 2.11, el trabajo que se efectúa sobre el sistema es
dV
V'<nb
La integración se realiza fácilmente, teniendo en cuenta que V - nb = x, y la
solución es
(
V2 - nb)Wrev = nRT In ---
VI-nb
= :...0.50x 8.3145 x 300(1) In [0.50 - 0.01)/(2.00 - 0.01)]
= 1247 (J) In (0.49/1.99) = 1247 (J) x 1.401
= 1.75 kJ
......'
~~.~: r-"
'•. f
'=. ,1
<f](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-96-320.jpg)




![86 Capítulo2 La primeraley de la termodinámica
2.39. Pruebe que para un gas ideal dos curvas adiabáticas re-
versibles de un diagrama P- V no se intersectan.
2.40. Un gas ideal es aquel que sigue la relación P V = nRT. En
la sección 2.7 se demostró que para este tipo de gases,
Pruebe que para un gas ideal, Cv y C» son independientes del
volumen y la presión.
2.41. Una mol de gas ideal se sometió a expansión isotérmica
reversible hasta que se duplicó'. Si el gas efectuó 1 kJ de trabajo,
¿cuál era su temperatura?
2.42. Un gas con comportamiento ideal se deja expandir de
manera adiabática y reversible hasta el doble de su volumen. Su
temperatura inicial era de 25.00°C, y Cv,m = (5/2)R. Calcule ~Um
y Mim para el proceso de expansión.
2.43. Con CV. m = (3/2)R, 1 mol de gas monoatómico ideal
experimenta un proceso reversible en el cual su volumen se
duplica; el gas absorbe 1 kJ de calor. La presión inicial es 1 bar
y la temperatura inicial, 300 K. El cambio de entalpía es 1.50 kJ.
a) Calcule la temperatura y la presión final.
b) Calcule ~Uy w para el proceso.
*2.44. Compruebe que
*2.45. Compruebe que para un gas ideal la tasa de cambio de la
presión dP/dt está relacionada con las tasas de cambio de volu-
men y temperatura como sigue:
1 dP 1 dV 1 dT
--=---+--
P dt V dt T dt
*2.46. Inicialmente 5 moles de nitrógeno se encuentran a 25°C
de temperatura y una presión de 10 bares. Puede suponerse que
el gas tiene comportamiento ideal; Cv.m = 20.8 J K-1
mol " y es
independiente de la temperatura. Suponga que la presión des-
ciende de manera repentina a 1 bar; calcule la temperatura final,
~Uy ~H.
2.47. Una reacción química se verifica a 300 K en una mezcla
gaseosa que se comporta de manera ideal, y la cantidad total de
gas aumenta 0.27 moles. Si ~U = 9.4 kJ, ¿cuál es el valor de ~ H?
2.48. Suponga que 1.00 mol de un gas monoatómico ideal [Cv
= (3/2)R] a 1 bar experimenta una compresión adiabática y
reversible de 0.1000 m3
a 0.0100 m3
a 25.0°C. Calculeq, w, ~U
y~H.
2.49. Suponga que un gas ideal experimenta un proceso adia-
bático isobárico irreversible. Derive expresiones para q, w, ~U
y ~ H, Yla temperatura final del gas que experimenta el proceso.
2.50. Exactamente 1 mol de gas monoatómico ideal a 25.0°C
se enfría y se expande de 1.00 dnr' a 10.00 dnr' contra una
presión externa de 1.00 bar. Calcule la temperatura final y q, W,
~Uy ~H.
2.51. Un globo de 15 m de diámetro se infla con helio a 20°C.
a) ¿Cuál será la masa de helio en el globo, suponiendo que el
gas es ideal?
b) ¿Cuánto uabajo efectúa el globo mientras se infla contra una
presión externa de 1 atm (101.315 kPa), de un volumen inicial
de cero hasta el volumen final?
2.52. a) Calcule el trabajo realizado cuando 1mol de un gas ideal
a una presión de 2 bares y 300 K se expande isotérrnicamente
hasta un volumen de 1.5 L, si la presión externa se mantie-
ne constante a 1.5 bares.
b) Suponga que ahora el gas se expande de manera isotérmica y
reversible hasta el mismo volumen final; calcule el trabajo
realizado ..
Gases reales
2.53. Para un gas ideal, PVm = RT y, por lo tanto, (dT/dP)v =
Vm/R.Derive la relación correspondiente para un gas de van der
Waals.
*2.54. Una mol de gas a 300 K se comprime de manera isotér-
mica y reversible desde un volumen inicial de 10 dnr' hasta un
volumen final de 0.2 dm', Calcule el trabajo realizado por el
sistema si:
a) El gas es ideal:
b)La ecuación de estado del gas eSP(Vm- b) = RT, con b = 0.03
dnr' mol ". Explique la diferencia entre los dos valores.
*2.55. Una mol de gas a 100 K se comprime isotérmicamente
de un volumen inicial de 20 dnr' a un volumen final de 5 dnr',
Calcule el trabajo realizado sobre el sistema si:
a) El gas es ideal.
b) La ecuación de estado es
(
p + -; Ir; = RT donde a = 0.384 m" Pa mol-I
vmJ
[Esta ecuación se sigue de manera aproximada a bajas tempera-
turas, mientras que la expresión P(Vm - b) = RT (véase el
problema 2.54) se sigue con más exactitud a temperaturas más
altas.] Explique la diferencia entre los valores obtenidos en a)
y b).](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-101-320.jpg)





![(3.1)
92 Capítulo 3 Segunda y tercera ley de la termodinámica
(es decir, no se permite que entre o salga calor del sistema); esto puede llevarse a cabo
rodeando el cilindro con material aislante. Como el gas efectúa trabajo durante la
expansión y no se le aporta calor, la temperatura debe descender; la temperatura final
se llamará Te Y la presión y el vólumen, P3 y V3, respectivamente. El tercer paso
implica comprimir el gas de manera isoténnica (a la temperatura Te) hasta que la
presión y el volumen sean P4 Y V4. Por último, el gas se comprime adiabáticamente
hasta que regresa a su estado original A (p¡, VI, Th)' Al efectuar trabajo sobre el
sistema sin transferencia de calor, la temperatura aumenta de Te a Th'
A continuación se considerarán con más detalle los cuatro pasos. En particular,
se desea determinar los valores de !1U para cada paso, la cantidad de calor absorbido
(q) y el trabajo realizado (w). Las expresiones para estas cantidades se resumen en la
tabla 3.1.
Expansión isotérmica
reversible
l. El paso A ~ B es la expansión isotérmica reversible a Ti: En la sección 2.7 se
comprobó que cuando se efectúa la expansión isoténnica de un gas ideal, no hay
cambio de energía interna:
También se demostró (Ec. 2.72) que el trabajo realizado sobre el sistema en un proceso
isotérmico reversible es RT In (V¡nicia¡/Vfina¡):
(para 1 mol) (3.2)
y como por la primera ley
(3.3)
se deduce que .
(3.4)
Expansión adiabática
reversible
2. En el paso B ~ ese rodea al cilindro de una camisa aislante y se permite que
el sistema se expanda de manera reversible y adiabática hasta el volumen V3.
Como el proceso es adiabático,
(3.5)
TABLA 3.1 Valores de /j,U, q y w para los cuatro pasos reversibles en el ciclo
de Carnot, para 1 mol de gas ideal.
Paso !1U
o
Cl! (Te - Th)
O
C; (Th - Te)
R Thln~
O
RT.ln~
e v]
O
Neto O R it,- Te) In 1-,
(porque 1-= j; véase la Ec. 3.16)
, ]](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-107-320.jpg)








![Prueba de que la entropía
total debe aumentar
en un proceso natural
Sistema aislado:
el proceso A--tB es irreversible
rBdq¡rr _ - - B
lA r=o,,-/'
/'
/
A
Sistema no aislado:
el proceso B--tA es reversible
l
A
dqrev
f1SB-.A = -- < O
B T
f1SA-.B> O
3.2 Procesosirreversibles 101
Este resultado, es decir, que el proceso espontáneo (y por lo tanto, irreversible)
se lleva a cabo con un aumento total de entropía en el sistema y los alrededores, es
universalmente válido. Su prueba se basa en el hecho de que la eficiencia del ciclo
de Carnot, en el cual algunos pasos son irreversibles, debe ser inferior a la de un
ciclo totalmente reversible, ya que los sistemas que experimentan procesos rever-
sibles efectúan trabajo máximo. Por tanto, en lugar de la ecuación 3.23 se tiene la
expresión para un ciclo irreversible,
q irr + q irr T - T
h c <_h __ e
qJ,rr r,
(3.39)
Esta relación se reduce a
q
irr qirr
_h_+_c_<O
t; t,
(3.40)
de manera que, en general, para cualquier ciclo que no sea completamente reversible,
(3.41)
Esto se conoce como desigualdad de Clausius, en honor de Rudolf Clausius, quien
sugirió dicha relación en 1854.
Considérese que el cambio irreversible del estado A al estado B es un sistema
aislado [Fig. 1.2c], que se representa mediante la línea punteada de la figura 3.6.
Supóngase que posteriormente se modifican las condiciones; el sistema ya no está
FIGURA 3.6 aislado. Por último: éste regresa a su estado inicial A por la trayectoria reversible
Proceso cíclico en dos etapas: 1) el representada por la línea continua de la figura 3.6. Durante este proceso reversible, el
sistema se encuentra aislado y cambia sistema no está aislado y puede intercambiar calor y trabajo con el medio ambiente.
del estado A al estado B mediante un Como todo el ciclo A ~ B ~ A es parcialmente reversible, se puede aplicar la
proceso irreversible (línea punteada); ecuación 3.41 lo cual significa que
2) el sistema no está aislado y cambia '
de estado mediante un proceso
reversible (línea continua). B dq, A dq
f ~+f ~<O
A T B T
(3.42)
La primera integral es igual a cero ya que el sistema estuvo aislado durante el proceso
irreversible, de manera que cualquier cambio de calor en una parte del sistema se
compensó exactamente con un cambio igual y opuesto en otra parte. La segunda
integral es el cambio de entropía cuando ocurre el proceso B ~ A, de modo que
(3.43)
En consecuencia, se deduce que
(3.44)
Por lo tanto, la entropía del estado final B es siempre mayor que la del estado inicial
A cuando el proceso A ~ B se lleva a cabo de manera irreversible en un sistema
aislado.](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-116-320.jpg)




















![122 Capítulo3 Segunday terceraleyde la termodinámica
Energía de Gibbs y trabajo reversible
Cuando un gas ideal se comprime reversiblemente a temperatura constante, el trabajo
que se efectúa es el incremento de energía de Gibbs. En la sección 2.7 se vio que el
trabajo reversible realizado para comprimir n moles de un gas ideal, a la temperatura
T, del volumen V] al volumen V2 es
V] P2
W =nRTln-=nRTln-rey . V
2
P,
(3.92)
Durante este proceso isotérmico no hay cambio de energía interna; la energía interna
de un gas ideal está únicamente en función de la temperatura, y no de la presión o
elvolumen. De la primera ley se deduce que el calor absorbido por el sistema es el
negativo del trabajo que se efectúa sobre el sistema:
V2
qrev = nRTIn -
V]
(3.93)
El cambio de entropía (que numéricamente es una disminución porque V] > V2) es,
por tanto,
A qrev 1 V2
uS=-=nR n-
T V]
(3.94)
No hay cambio de entalpía; la entalpía para un gas ideal está en función únicamente
de la temperatura. Entonces el cambio de energía de Gibbs es
IJ.G = IJ.H - Té: S (3.95)
V] P2
=nRTln - = nRTln-
V2
r, (3.96)
En consecuencia, el trabajo reversible efectuado sobre el sistema (Ec. 3.92) es el
cambio de su energía de Gibbs.
Una relación aún más importante entre la energía de Gibbs.y el trabajo surge para
procesos que ocurren a temperatura y presión constantes. El trabajo se puede clasifi-
car en dos tipos: trabajo que surge por un cambio de volumen que se presenta cuando
tiene lugar un proceso, y cualquier otro tipo de trabajo. Por ejemplo, cuando funciona
una celda electroquímica puede haber un pequeño cambio de volumen; se efectúa un
trabajo sobre los alrededores o éstos efectúan trabajo sobre el sistema. Sin embargo,
de mucho mayor interés e importancia práctica es el trabajo eléctrico que se obtiene
cuando la celda funciona. El trabajo que surge por el cambio de volumen se denomina
trabajo PV y se representa con el símbolo Wpv, Cualquier otro tipo de trabajo se
llamará trabajo no-PV, Wno-PV' Por tanto, el trabajo total es
W = Wpv + wno-pv (3.97)
Otro tipo de trabajo no-PV es el trabajo osmótico. El trabajo no-PV se llama en
ocasiones trabajo net.o.
Ahora se derivará el importante resultado de que el trabajo no-PV es igual al
cambio de energía de Gibbs para un proceso reversible que ocurra a temperatura y
presión constantes. Se comienza con la definición de la energía de Gibbs (Ec. 3.80):
G = H - TS= U+ PV - TS (3.98)](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-137-320.jpg)

![124 Capítulo3 Segunday terceraley de la termodinámica
u
FIGURA 3.11
Dispositivo mnem6nico para obtener
las ecuaciones 3.116-3.119 Y las
relaciones de Maxwell 3.122-3.125.
Cada uno de los cuatro potenciales
termodinámicos U, A, G Y H, está
f1anqueado por dos propiedades (p. ej.,
U está f1anqueado por V y S) con las
cuales tiene una relación especial. El
sentido de la flecha indica el signo de
la ecuaci6n.
a) Para las ecuaciones 3.116-3.119,
cualquier potencial termodinámico
puede diferenciarse con respecto a
una de las propiedades vecinas si se
mantiene la otra constante. El
resultado se obtiene siguiendo la
flecha. Por ejemplo,
(
aUl = T Y (dU) =_p
as L av s
b) Para las ecuaciones 3.122 y 3.125,
cualquier derivada parcial de una
propiedad con respecto a una
propiedad vecina (p. ej., (dVIdT)p) se
relaciona con la correspondiente
derivada del otro lado del cuadrado [o
sea, (aS¡ap)r)]; las flechas indican los
signos (en este ejemplo, el signo es
negativo).
Aplicando la relación d(PV) = PdV + VdP, se efectúa la transformada de Legendre,
que se llama así en honor del matemático francés Adrien Marie Legendre (1752-
1853).
De manera semejante, para las energías de Gibbs y de Helmholtz se tiene
dA = d (U - T S) = dU - d (T S) = dU - T dS - S dT
= -P dV + T dS - T dS - S dT = -P dV - S dT
(3.1 08)
(3.109)
y
dG = d (H - T S) = dH - T dS - S dT
= V dP + T dS - T dS - S dT = V dP - S dT
(3.110)
(3.111)
Ahora se pueden combinar las expresiones obtenidas para dU, dH, dA Y dG con
relaciones generales del cálculo diferencial:
dU= -P dV + T dS = (au] dV + (au] dS
léW s las v
dH = V dP + T dS = (aHJ dP + (aH) dS
lap s las p
dA = -P dV - S dT = (aA] dV + (aA] dT
lav T let v
dG = V dP -S dT= (dGJ dP + (dG] dT
ldP T ldT p
(3.112)
(3.113)
(3.114)
(3.115)
y se obtienen relaciones importantes igualando los coeficientes; así, de la ecuación
3.112 se tiene
(
dU) =_P
dV s
(
dU)- -T
dS v
(3.116)
De manera similar, de las ecuaciones 3.113, 3.114 Y 3.115,
(
dH)- -V
ap s (
dH)- -T
as p
(3.117)
(dA) =_P
dV r
(3.118)
(3.119)
En la figura 3.11 se ilustra un aparato mnemónico para obtener estas ocho relaciones ..](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-139-320.jpg)

![126 Capítulo 3 Segunda y tercera ley de la termodinámica
Ecuación termodinámica
de estado
(3.126)
(3.127)
Entonces, según las ecuaciones 3.118 Y 3.124 la ecuación termodinámica de estado
para U es
(3.128)
La ecuación termodinámica de estado correspondiente para H se obtiene como
sigue:
(
OHJ = [O(G + T S)] = (OGJ + T (OSJ
oP T P T oP T oP T
Y empleando las ecuaciones 3.119 Y 3.125 se obtiene la ecuación termodinámica de
estado para H,
(3.129)
(OHJ (OVJ- -v T-
oP T st p
(3.130)
Algunas aplicaciones de las relaciones termodinámicas
Se pueden obtener muchas otras relaciones entre las cantidades termodinámicas
basándose en las ecuaciones derivadas en estas últimas páginas. A continuación se
dan algunos ejemplos.
Una aplicación de utilidad se relaciona con la teoría del efecto de Joule- Thomson,
que se consideró en la sección 2.8. El coeficiente /-t de Joule- Thomson se define según
la ecuación 2.108:
(3.131)
y Cp se define por medio de la ecuación 2.27:
e,== (OHJ
oT p
Sin embargo, según la teoría de las derivadas parciales (apéndice C),
(3.132)
(3.133)
de manera que
(3.134)](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-141-320.jpg)



![130 Capítulo 3 Segunda y tercera ley de la termodinámica
Área = RTln(f2/P2); (12< P2)
(en este caso es negativa)
!tIa..
,
-::.E
P, =0 P2
Presión, P
FIGURA 3.13
Diagrama esquemático de Vm - RTIP
para 1 mol dé gas real.
Si P, es una presión suficientemente baja,./]/P, es igual a la unidad, de manera que
RT In h = f2(Vm
_ RT)dP
P2 P, P
Supóngase, por ejemplo, que se cuenta con datos confiables de presión y volumen
que abarcan una amplia gama de presiones y descienden hasta presiones muy bajas,
a una temperatura T dada. Entonces se puede calcular (Vm - RT)/P y graficar esta
cantidadcontraP, como se muestra en el diagrama de la figura 3.13. Después se puede
obtener el valor del área sombreada en la figura 3.13, desde P, = O hasta cualquier
valor P2. Esta área es la integral del lado derecho de la ecuación 3.158, y permite
calcular la fugacidad h. Se han utilizado diversos procedimientos analíticos para
evaluar estas integrales. En ocasiones se aplica la ley de los estados correspondientes
(Sección 1.12); mientras esta ley sea válida, todos los gases se adaptan a un mismo
conjunto de curvas, y para un gas dado sólo es necesario conocer sus constantes
.críticas.
EJEMPLO 3.9 Cuando el oxígeno se encuentra a presiones que no son
demasiada altas sigue la ecuación
P(Vm-b)=RT
donde b = 0.0211 dnr' mol"'
a) Calcule la fugacidad del oxígeno gaseoso a 25°C y presión de 1 bar.
b) ¿A qué presión la fugacidad será igual a 1 bar?
Solución Para esta ecuación de estado
·RT
V --=bm P
de manera que
a) Por tanto, la ecuación 3.159 da, con los datos P¡ = O,P2 = 1 bar y b= 0.0211
dnr' mol ", da
RT lnh = 0.0211 bar dnr' mol"'
= 0.0211 x 105
x 10-3 Pa m3 mol"'
0.0211 x 102
(Pa m3
mol'")
In!: - -----,---,---_._.........
2 - 8.3145(J K-1 mor') x 298.15K
= 8.51 x 10-4
h = 1.0009 bar
bP
b) Inf=lnP+-
RT
En consecuencia, la presión a la cualf= 1 bar (lnf= O)es
bP
In P = - -- o P = e-bPIRT
RT
(3.159)](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-145-320.jpg)

![132 Capítulo 3 Segunda y tercera ley de la termodinámica
de manera que el lado izquierdo de la ecuación 3.164 puede expresarse como
T [~(I1G]aT T p
Por tanto,
T [~(I1G] I1H
aT T p T
(3.166)
o
(3.167)
Esta importante relación termodinámica se conoce como ecuación de Gibbs-Helm-
holtz. Si los reactivos y productos se encuentran en sus estados estándar, la ecuación
adopta la forma
(3.168)
Esta ecuación es de particular importancia al considerar cómo dependen las constan-
tes de equilibrio de la temperatura (Sección 4.8).
3.10 LIMITACIONES TERMODINÁMICAS PARA LA TRANSFORMACiÓN
DE ENERGíA
Las leyes de la termodinámica tienen muchas aplicaciones prácticas a la interconver-
sión de diversos tipos de energía; esto constituye un problema de creciente importan-
cia técnica y económica. Tanto la primera como la segunda leyes ponen límites a la
cantidad de energía o trabajo útil que se puede obtener de determinada fuente.
Eficienc:ias de la primera ley
La primera ley se refiere simplemente al principio de conservación de la energía, y
su aplicación es muy directa; una energía que no sirva para el propósito que se desea,
se debe restar del total para obtener la cantidad de energía útil. La eficiencia con la
cual la energía contenida en un combustible se transforma en energía útil es muy
variable.' Cuando se efectúa la combustión de madera o carbón en una chimenea,
aproximadamente el 80% del calor escapa por el tiro; sólo un 20% permanece en la
habitación. Por otra parte, una chimenea doméstica de buena calidad puede transfor-
mar cerca del 75% de la energía del combustible en calor útil. Sin embargo, muchas
chimeneas domésticas funcionan a eficiencias inferiores, cercanas al 50 o 55%. Estas
7 Los valores característicos de la eficiencia pueden encontrarse en un artículo de C. M. Summers,
Scientific American. 225, 149 (septiembre de 1971).](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-147-320.jpg)











![*3.43. Inicialmente a 300 K Ypresión de 10 atm, 1 mol de gas
se expande adiabáticamente contra una presión constante de
4 atm hasta alcanzar el equilibrio. Suponga que el gas es ideal
con
CP. m/J K-I
mol-I
= 28.58 + 1.76 x 10-2
T/K
y calcule 1::1u, 1::1 H Y1::1 S.
3.44. Calcule 1::1 H~ I::1GoY1::1 So para la reacción
CH4 (g) + 202 (g) -7 CO2 (g) + 2H20 (1)
empleando los datos del apéndice D.
3.45. El siguiente es un conjunto de condiciones especiales:
a) Válido únicamente para un gas ideal.
b) Válido únicamente para un proceso reversible.
e) Válido únicamente cuando S es la entropía total (sistema +
alrededores ).
d) Válido únicamente para un proceso isotérmico que ocurra a
presión constante.
e) Válido únicamente para un proceso isotérmico que ocurra a
volumen constante.
Considere cada una de las siguientes afirmaciones e indique
cuáles de las condiciones anteriores deben aplicarse para que di-
cha afirmación sea cierta:
a) 1::1U = Opara un proceso isotérmico.
b) L H = Opara un proceso isotérmico.
e) L S total = Opara un proceso adiabático.
d) L S > Opara un proceso espontáneo.
e) LG < Opara un proceso espontáneo.
3.46. Calcule los cambios de energía de Gibbs y la entropía para
la transformación de 1 mol de agua líquida a 100°C y presión de
1 bar en vapor a la misma temperatura y presión de 0.1 bares.
Suponga comportamiento ideal. El calor de evaporación del
agua a 100°C es 40.6 kJ mol ".
3.47. La bacteria nitrobacter efectúa la siguiente reacción:
NOZ" + ~02 -7 NO~
Utilice los datos del apéndice D para calcular L H~ GO Y L So
para la reacción.
Transformación de energía
3.48. A una presión de 100 atm el agua hierve a 312°C, mientras
que a 5 atm hierve a 152°C. Compare las eficiencias de Camot
de motores de vapor a 100 atm y 5 atrn si Te es 30°C.
Problemas 143
3.49. Se diseña un sistema de enfriamiento para mantener un
refrigerador a -4°C en una habitación a 20°C. Si se fugan 104
J
de calor del refrigerador por minuto y el sistema funciona al
40% de la eficiencia termodinámica máxima. ¿qué potencia
necesita en watts? [1 watt (W) = 1 J S-l.]
3.50. Se emplea una bomba calorifica para mantener la tempe-
ratura de una casa a 25°C. Calcule el factor de desempeño
máximo de la bomba cuando la temperatura externa es a) 20°C,
b) O°C y e) -20°C.
3.51. Un motor de automóvil común funciona con una tempe-
ratura de 2000°C en los cilindros y una temperatura de salida de
800°C. Un combustible de octanaje representativo (masa mo-
lar = 114.2 g mol ") tiene una entalpía de combustión de -5500
kJ mol"' y 1 dm" (0.264 U.S. gal) tiene una masa de 0.80 kg.
Calcule la cantidad máxima de trabajo que puede efectuar la
combustión de 10 dnr' de dicho combustible.
3.52. La temperatura de un edificio se mantiene a 20°C median-
te una bomba de calor, y cierto día la temperatura externa es de
10°C. Se aporta trabajo a la bomba calorífica mediante un motor
de calor que quema combustible a 1000°C y funciona a 20°C.
Calcule el factor de desempeño del sistema (o sea, la relación
entre el calor que se aporta al edificio y el calor que produce el
combustible del motor de calor). Suponga eficiencias perfectas
en la bomba y el motor.
3.53. Suponga que un refrigerador enfria a O°C, descarga calor
a 25°C y funciona con una eficiencia del 40%.
a) ¿Cuánto trabajo se requeriría para congelar 1 kg de agua
(I::1¡H= -6.02 kJ mol-I
)?
b) ¿Cuánto calor se descargaría durante el proceso?
Relaciones termodinámicas
*3.54. Suponga que un gas sigue la ecuación de van der Waals
(
p + ";J (Vm - b) = RT
Vm
Demuestre que
*3.55. Obtenga una expresión para el coeficiente de Joule-
Thomson para un gas que sigue la siguiente ecuación de estado
P(V •• -b) =RT
en términos de R, T. P, Vm y CP.m'](https://image.slidesharecdn.com/212974199-fisicoquimica-laidler-170612014728/85/212974199-fisicoquimica-laidler-159-320.jpg)

El documento presenta una segunda edición en español de un libro de texto sobre fisicoquímica. Se trata de una traducción de la segunda edición en inglés y contiene 20 capítulos que cubren diversas áreas de la fisicoquímica como termodinámica, equilibrio químico, cinética química y espectroscopia. El libro fue traducido al español y revisado por expertos para su uso en cursos universitarios de fisicoquímica.



































































