También conocidoscomo haloalcanos, son
compuestos orgánicos que contienen halógenos
unidos a un átomo de carbono saturado con
hibridación sp3
.
Los halogenuros de alquilo son compuestos derivados
de los alcanos , que pueden tener uno o mas
halógenos; existen 3 clases principales de compuestos
orgánicos halogenados que son :Halogenuros de
alquilo , vinilo y arilo.
Las moléculas de los halógenuros de alquilo son
relativamente abundantes en la naturaleza y en la
industria, los cuales tienen deferentes aplicaciones.
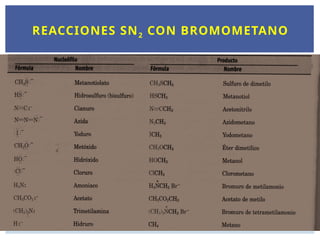
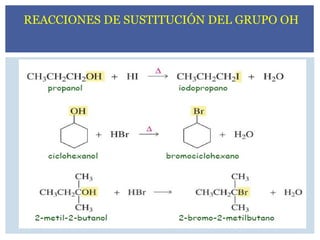
HALOGENUROS DE ALQUILO
3.
CH3Br Cl2CH2 CF3-CHClBr
Bromurode metilo Diclorometano halotano
(pesticida) (solvente) (anestésico)
Derivado de triazol
(fungicida)
HALOGENUROS DE ALQUILO
4.
En la nomenclaturade la IUPAC, el halógeno se
considera como un sustituyente, de la cadena
carbonada.
REGLAS
1.Se identifica la cadena principal.
2.Se enumeran los C por el extremo mas cercano al
primer sustituyente, sin importar si se trata de un
alquilo o de un halógeno.
3.Se nombran los sustituyentes en orden alfabético y
finalmente la cadena principal.
NOMENCLATURA
5.
4. Citar enprimer lugar el X2 seguido del nombre del
hidrocarburo, indicando la posición que ocupa el X2 en
la cadena, sabiendo que el doble y triple enlace, tienen
prioridad sobre el X2 al enumerar la cadena.
Cl-CH2 -CH2 -CH2 -CH3 ( 1-clorobutano)
5. Si aparece el mismo X2 repetido, se utilizan los
prefijos di, tri, tetra
CH2=CH-CCl2- CH2Cl (3,3,4-tricloro-1-buteno)
6. Cuando todos los H2 de un hidrocarburo están
sustituidos por un X2, se anteponen el prefijo per al
nombre del halógeno.
CCl3-CCl2-CCl2-CCl2CCl3 (percloropentano)
NOMENCLATURA
6.
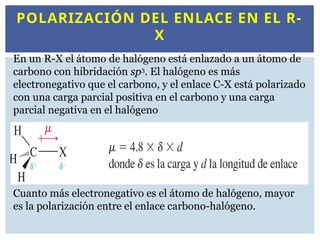
POLARIZACIÓN DEL ENLACEEN EL R-
X
En un R-X el átomo de halógeno está enlazado a un átomo de
carbono con hibridación sp3
. El halógeno es más
electronegativo que el carbono, y el enlace C-X está polarizado
con una carga parcial positiva en el carbono y una carga
parcial negativa en el halógeno
Cuanto más electronegativo es el átomo de halógeno, mayor
es la polarización entre el enlace carbono-halógeno.
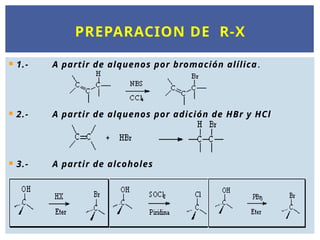
7.
1.- Apartir de alquenos por bromación alílica.
2.- A partir de alquenos por adición de HBr y HCl
3.- A partir de alcoholes
PREPARACION DE R-X
8.
Los R-X seconvierten fácilmente en otros grupos
funcionales. El átomo de X2 puede salir con su par de
electrones de enlace para formar un ión haluro estable;
se dice que un haluro es un buen grupo saliente.
Cuando otro átomo reemplaza al ión haluro, la
reacción es una sustitución. En una reacción de SN, el
X-
se sustituye por un Nu-
Si el X-
abandona la molécula junto con otro átomo o
ion, produciendo un alqueno la reacción es una
eliminación. En esta reacción el X-
se "elimina" de la
molécula después de la abstracción de un H2 por
medio de una BF.
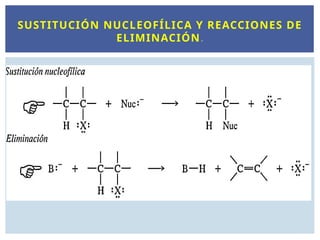
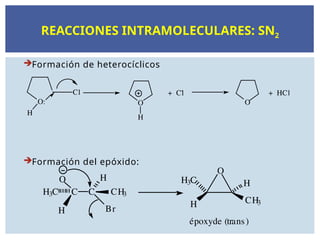
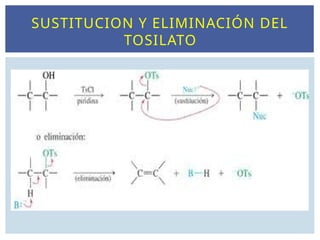
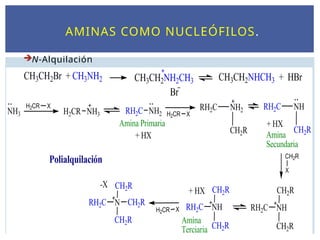
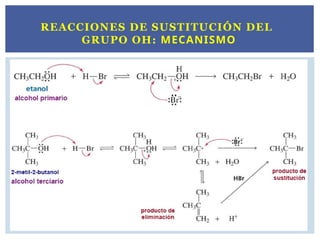
SUSTITUCIÓN NUCLEOFÍLICA Y
REACCIONES DE ELIMINACIÓN.
Las reacciones deSN transcurren generalmente sobre
los R-X.
Estos compuestos están polarizados en el enlace del
halógeno generando un carbono electrófilo.
Los que sustituyen el X2 en el enlace C-X son los
nucleófilos.
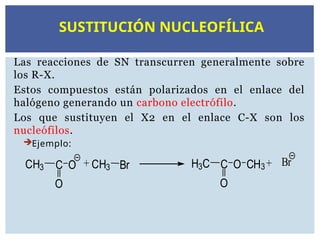
Ejemplo:
CH3 C
O
O + CH3 Br H3C C
O
O CH3+ Br
SUSTITUCIÓN NUCLEOFÍLICA
11.
SUSTITUCIÓN NUCLEOFÍLICA
Un nucleófilopuede ser neutro « Nu » (una
molécula, o grupo funcional con un par
libre de electrones) o un anión (Nu-
).
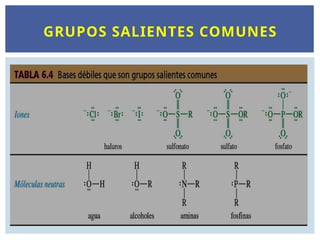
X es el grupo saliente, los buenos grupos
salientes son bases débiles. Ejemplo los
iones halogenuros.
Principalmente existen dos mecanismos de
sustitución: El mecanismo SN1 y el
mecanismo SN2.
neutro
12.
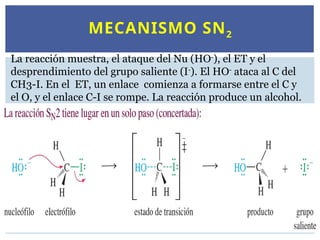
MECANISMO SN2
La reacciónmuestra, el ataque del Nu (HO-
), el ET y el
desprendimiento del grupo saliente (I-
). El HO-
ataca al C del
CH3-I. En el ET, un enlace comienza a formarse entre el C y
el O, y el enlace C-I se rompe. La reacción produce un alcohol.
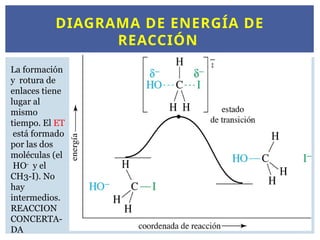
13.
DIAGRAMA DE ENERGÍADE
REACCIÓN
La formación
y rotura de
enlaces tiene
lugar al
mismo
tiempo. El ET
está formado
por las dos
moléculas (el
HO-
y el
CH3-I). No
hay
intermedios.
REACCION
CONCERTA-
DA
14.
La reacción esdel orden 2 (1 respecto a RX y 1
respecto Nu-
) SN2 (sustitución nucleofílica de
segundo orden o bimolecular). Esto se debe a que
en el estado de transición interactúan el
halogenuro y el nucleófilo, por lo que se dice que
la velocidad es función de RX y de Nu-
v = k[RX][Nu-
]
En consecuencia esta reacción tiene una cinética
de segundo orden.
CINETICA DE LA REACCION SN2
Para formarun enlace, el nucleófilo tiene que
encontrarse dentro de la distancia enlazante del
carbono. Un haluro de alquilo estéricamente
impedido evitará que el nucleófilo se acerque lo
suficiente como para reaccionar.
El ataque SN2 en un haluro de alquilo primario
sencillo no está impedido; el ataque en un haluro de
alquilo secundario está impedido, y el ataque en un
haluro de alquilo terciario es imposible
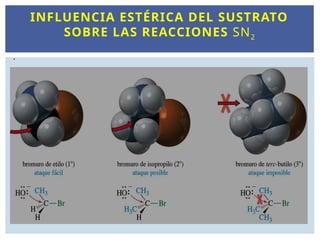
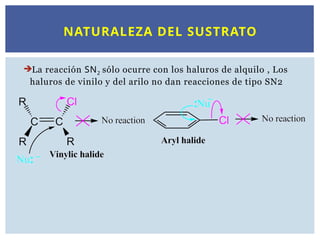
ESTEREOQUIMICA DE LA SN2 : INFLUENCIA
ESTÉRICA DEL SUSTRATO
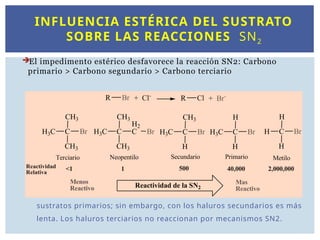
INFLUENCIA ESTÉRICA DELSUSTRATO
SOBRE LAS REACCIONES SN2
El impedimento estérico desfavorece la reacción SN2: Carbono
primario > Carbono segundario > Carbono terciario
La reacción transcurre con rapidez en los haluros de metilo y con los
sustratos primarios; sin embargo, con los haluros secundarios es más
lenta. Los haluros terciarios no reaccionan por mecanismos SN2.
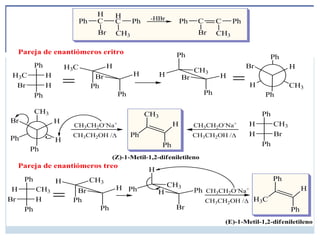
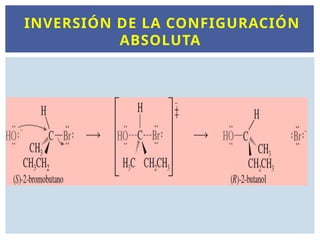
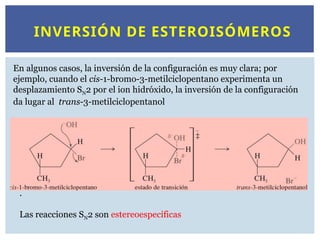
INVERSIÓN DE ESTEROISÓMEROS
Enalgunos casos, la inversión de la configuración es muy clara; por
ejemplo, cuando el cis-1-bromo-3-metilciclopentano experimenta un
desplazamiento SN2 por el ion hidróxido, la inversión de la configuración
da lugar al trans-3-metilciclopentanol
.
Las reacciones SN2 son estereoespecíficas
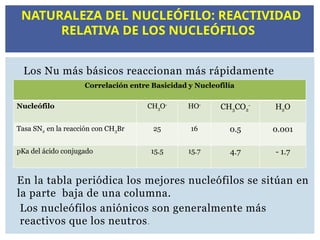
Cuanto más fuertesea el Nu más favorecida estará la
reacción SN2.Los Nu son, en general, BL neutras o
cargadas negativamente.
NATURALEZA DEL NUCLEÓFILO
Los Nu másbásicos reaccionan más rápidamente
En la tabla periódica los mejores nucleófilos se sitúan en
la parte baja de una columna.
Los nucleófilos aniónicos son generalmente más
reactivos que los neutros.
Correlación entre Basicidad y Nucleofilía
Nucleófilo CH3O-
HO-
CH3CO2
-
H2O
Tasa SN2 en la reacción con CH3Br 25 16 0.5 0.001
pKa del ácido conjugado 15.5 15.7 4.7 - 1.7
NATURALEZA DEL NUCLEÓFILO: REACTIVIDAD
RELATIVA DE LOS NUCLEÓFILOS
27.
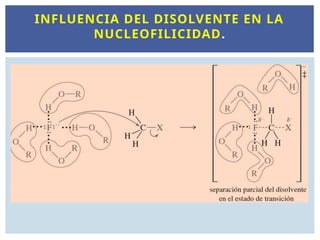
En un disolventeprótico el nucleófilo, disminuye su
fuerza
Si el ion es más pequeño, como el fluoruro, se
requiere más energía para poder separar el
disolvente de este ion que está fuertemente solvatado
que de otro ion más grande, que está más débilmente
solvatado, como el yoduro
Los disolventes próticos polares rodearán al
nucleófilo y reducirán su nucleofilia. Cuanto más
pequeño sea el átomo, más solvatado estará y mayor
será la reducción de la reactividad.
INFLUENCIA DEL DISOLVENTE EN LA
NUCLEOFILICIDAD.
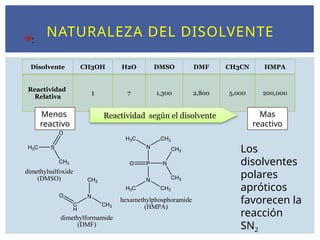
NATURALEZA DEL DISOLVENTE
:
S
O
CH3
H3C
dimethylsulfoxide
(DMSO)
C
H
ON
CH3
CH3
dimethylformamide
(DMF)
P
O
N
N
N
H3C CH3
CH3
CH3
CH3
H3C
hexamethylphosphoramide
(HMPA)
Disolvente CH3OH H2O DMSO DMF CH3CN HMPA
Reactividad
Relativa
1 7 1,300 2,800 5,000 200,000
Reactividad según el disolvente
Menos
reactivo
Mas
reactivo
Los
disolventes
polares
apróticos
favorecen la
reacción
SN2
30.
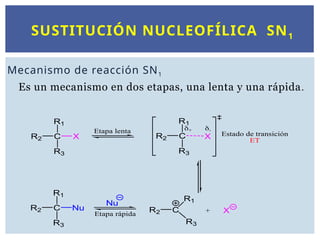
SUSTITUCIÓN NUCLEOFÍLICA SN1
Mecanismode reacción SN1
Es un mecanismo en dos etapas, una lenta y una rápida.
R1
C
R3
R2 X
R1
C
R3
R2 X
+ -
Estado de transición
ET
R1
C
R3
R2 + X
R1
C
R3
R2 Nu
Nu
Etapa rápida
Etapa lenta
La velocidad dereacción depende sólo de la etapa
lenta y por lo tanto sólo de la concentración del
sustrato RX y no de la del nucleófilo Nu-
.
v = k[RX]
La ecuación de velocidad es de primer orden: primer
orden respecto a la concentración del haluro de alquilo
y de orden cero respecto a la concentración del
nucleófilo.
CINETICA DE LA REACCION SN1
33.
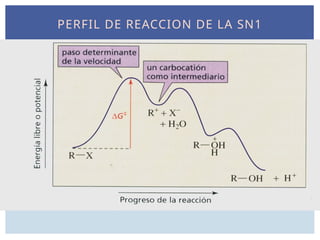
PERFIL DE REACCIONDE LA SN1
ET1
ET2
C X
+
+ Nu-
C
Nu
+
+ X-
C X-
+ Nu-
+
G
C X + Nu-
Carbocation
C
Nu + X-
Estado inicial
Estado final
LA REACCIÓN SN1DEPENDE DE LA
ESTABILIDAD DE CARBOCATIÓN
El paso limitante de la velocidad de la
reacción SN1 es la ionización para formar
un carbocatión, proceso fuertemente
endotérmico. El estado de transición
para este proceso endotérmico se
asemeja al carbocatión, por lo que las
velocidades de las reacciones SN1 tienen
una dependencia de la estabilidad del
carbocatión.
36.
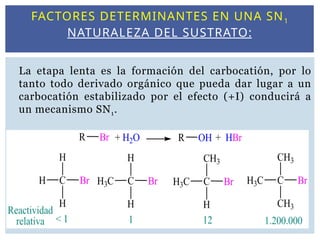
FACTORES DETERMINANTES ENUNA SN1
NATURALEZA DEL SUSTRATO:
La etapa lenta es la formación del carbocatión, por lo
tanto todo derivado orgánico que pueda dar lugar a un
carbocatión estabilizado por el efecto (+I) conducirá a
un mecanismo SN1.
37.
H
H
H Br
H H
Bromurode alilo
H
H
H
H
H
H
H
H
H
H
H
H
H
H H
OH
OH-
Carbocation estabilizado por resonanacia
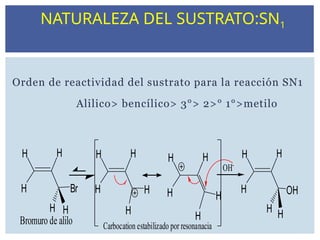
NATURALEZA DEL SUSTRATO:SN1
Orden de reactividad del sustrato para la reacción SN1
Alilico> bencílico> 3°> 2>° 1°>metilo
38.
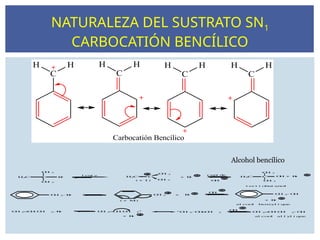
NATURALEZA DEL SUSTRATOSN1
CARBOCATIÓN BENCÍLICO
C
H 3
C
H 3
B
r
H3C H3C C
C
H 3
C
H 3
H3C C
C
H 3
C
H 3
O
H + B
r
C
H 2- O
H
C
H 2 + B
r
C
H 2- B
r
l en
t e
>
t er t i ob
ut anol
+ B
r
O
H
C
H 2=
C
H
- C
H 2- B
r
al coo
l benzyl i qu
e
C
H 2=
C
H
- C
H 2
+ +
C
H 2- C
H
=
C
H 2 C
H 2=
C
H
- C
H 2- O
H
O
H
al coo
l al l y
l i que
r ap
i d
e
C
O
H
+ B
r
( + I )
( + M)
+ B
r
Alcohol bencílico
39.
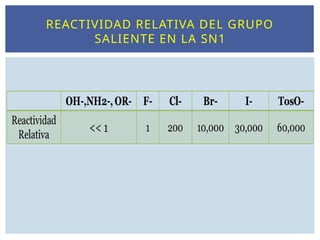
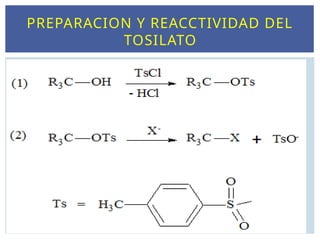
Cuanto más débilsea el enlace C-X más favorecida está la
formación del carbocatión (X debe atraer los electrones ).
Los mejores grupos salientes son bases débiles.
Los mejores grupos salientes son los iones más
voluminosos o capaces de estabilizar la carga negativa.
En medio ácido el OH de un alcohol se protona y el grupo
saliente es el H2O, que es mejor grupo saliente que el
halógeno.
El tosilato es un excelente grupo saliente (TsO-
).
NATURALEZA DEL GRUPO SALIENTE
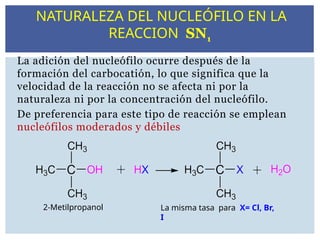
La adición delnucleófilo ocurre después de la
formación del carbocatión, lo que significa que la
velocidad de la reacción no se afecta ni por la
naturaleza ni por la concentración del nucleófilo.
De preferencia para este tipo de reacción se emplean
nucleófilos moderados y débiles
2-Metilpropanol La misma tasa para X= Cl, Br,
I
NATURALEZA DEL NUCLEÓFILO EN LA
REACCION SN1
44.
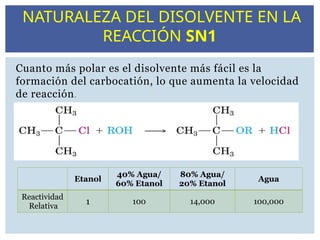
Cuanto más polares el disolvente más fácil es la
formación del carbocatión, lo que aumenta la velocidad
de reacción.
Etanol
40% Agua/
60% Etanol
80% Agua/
20% Etanol
Agua
Reactividad
Relativa
1 100 14,000 100,000
NATURALEZA DEL DISOLVENTE EN LA
REACCIÓN SN1
45.
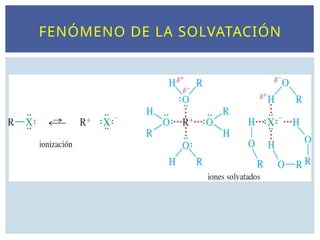
DISOLUCIÓN DE LOSIONES
La reacción SN1 está favorecida en disolventes polares, que
estabilizan los iones intermedios. El paso limitante de la
velocidad forma dos iones y la ionización tiene lugar en el
estado de transición. Los disolventes polares solvatan estos
iones debido a la interacción de los dipolos del disolvente con la
carga del ión. Los disolventes próticos como los alcoholes y el
agua son incluso disolventes más efectivos, ya que los aniones
forman enlaces de hidrógeno con el átomo de hidrógeno del
grupo -OH y los cationes, complejos con los electrones no
enlazantes del átomo de oxígeno del grupo -OH. Los disolventes
polares próticos, como los alcoholes, pueden solvatar mejor los
iones formados durante una reacción SN1.
NATURALEZA DEL DISOLVENTE
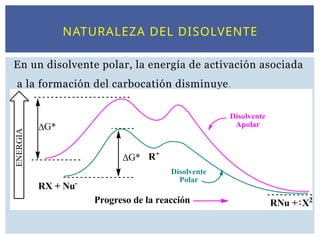
Enun disolvente polar, la energía de activación asociada
a la formación del carbocatión disminuye.
48.
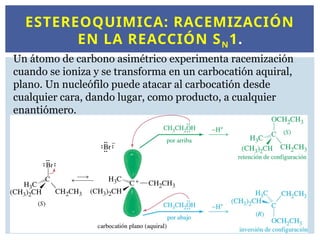
ESTEREOQUIMICA: RACEMIZACIÓN
EN LAREACCIÓN SN1.
Un átomo de carbono asimétrico experimenta racemización
cuando se ioniza y se transforma en un carbocatión aquiral,
plano. Un nucleófilo puede atacar al carbocatión desde
cualquier cara, dando lugar, como producto, a cualquier
enantiómero.
49.
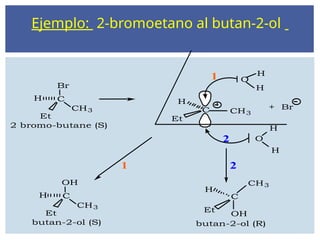
Ejemplo: 2-bromoetano albutan-2-ol
Br
C
CH3 C CH3
O
H
Et
1
2
H
Et
2 bromo-butane (S)
OH
C
CH3
H
Et
butan-2-ol (S)
O
H
H
1 2
C
CH3
Et
H
OH
butan-2-ol (R)
+ Br
H
H
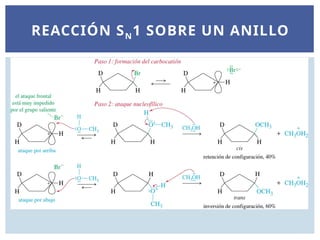
TRANSPOSICIÓN DE HIDRUROEN UNA
REACCIÓN SN1.
El producto reordenado, 2-etoxi-2-metilbutano, es el resultado de una
transposición de hidruro: movimiento de un átomo de hidrógeno con
su par de electrones de enlace. Una transposición de hidruro se
representa por el símbolo ~H. En este caso, la transposición de hidruro
convierte el carbocatión secundario inicialmente formado en un
carbocatión terciario más estable. El ataque del disolvente a este
último da lugar al producto de reordenamiento.
52.
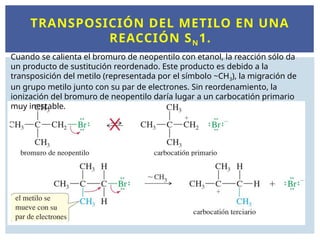
TRANSPOSICIÓN DEL METILOEN UNA
REACCIÓN SN1.
Cuando se calienta el bromuro de neopentilo con etanol, la reacción sólo da
un producto de sustitución reordenado. Este producto es debido a la
transposición del metilo (representada por el símbolo ~CH3), la migración de
un grupo metilo junto con su par de electrones. Sin reordenamiento, la
ionización del bromuro de neopentilo daría lugar a un carbocatión primario
muy inestable.
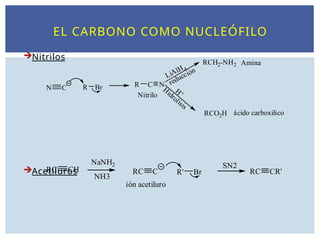
EL CARBONO COMONUCLEÓFILO
Nitrilos
Acetiluros
N C R Br R C N
Nitrilo
LiAlH 4
reducción
RCH2-NH2
H +
Hidrolisis
RCO2H
Amina
ácido carboxilico
R' Br
RC CH
NaNH2
NH3
RC C
ión acetiluro
RC CR'
SN2
58.
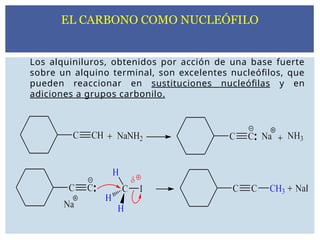
Los alquiniluros, obtenidospor acción de una base fuerte
sobre un alquino terminal, son excelentes nucleófilos, que
pueden reaccionar en sustituciones nucleófilas y en
adiciones a grupos carbonilo.
EL CARBONO COMO NUCLEÓFILO
59.
Sustrato: alílico> bencílico > alquilo terciario > alquilo
secundario > alquilo primario
Nucleófilo: no afecta la reacción
Grupos salientes: Los buenos grupos salientes (bases
débiles) favorecen la reacción
Disolvente: Los disolventes polares favorecen la reacción
por estabilización del carbocatión.
Estereoquímica: racemización
RESUMEN DE SN1
60.
COMPARACIÓN SN1 YSN2
SN1 SN2
Factores que influyen
Nucleófilo
Son apropiados los nucleófilos
débiles
Se necesitan nucleófilos
fuertes
Sustrato (RX) 3º > 2º CH3X > 1º > 2º
Disolvente
Se necesitan disolventes
ionizantes buenos
Amplia variedad de
disolventes
Grupo Saliente Ha de ser bueno Ha de ser bueno
Características
Cinéticas Primer orden kr[RX]
Segundo orden, kr[RX]
[Nu-
]
Estereoquímica
Mezcla de inversión y
retención
Inversión completa
Transposiciones Común Imposible
61.
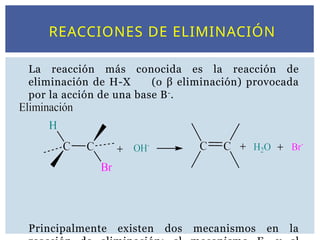
REACCIONES DE ELIMINACIÓN
Lareacción más conocida es la reacción de
eliminación de H-X (o β eliminación) provocada
por la acción de una base B-
.
Principalmente existen dos mecanismos en la
62.
Una eliminaciónimplica la pérdida de dos átomos o
grupos del sustrato, generalmente con la formación
de un enlace pi. Dependiendo de los reactivos y de
las condiciones en las que se encuentren, una
eliminación debería ser un proceso de primer orden
(E1) o de segundo orden (E2). Los siguientes
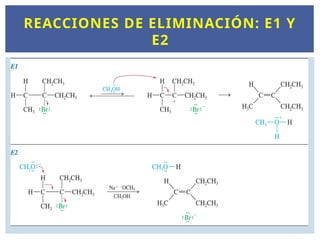
De la misma forma que existe un SN1 y un SN2,
existen dos mecanismos de eliminación, E1 y E2.
REACCIONES DE ELIMINACIÓN: E1 Y E2.
Al igualque el SN1, la reacción E1 implica
carbocationes intermedios.
El primer paso es la ionización de la molécula para
formar el carbocatión, seguido de la abstracción de
un protón del carbono cercano. El producto principal
de las reacciones de eliminación son los alquenos.
Los electrones que antes formaban el enlace carbono-
hidrógeno ahora forman el enlace pi entre dos
átomos de carbono. El mecanismo general de una
reacción E1 es:
MECANISMO E1
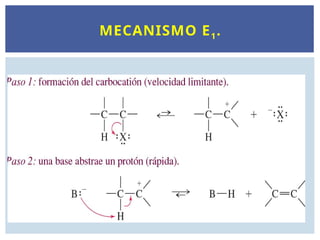
El primerpaso de los dos mecanismos es el mismo:
formación del intermedio carbocatiónico. El alcohol
puede actuar como una base o como un nucleófilo.
Si actúa como una base, la abstracción de un protón
producirá un alqueno. Si el alcohol actúa como un
nucleófilo, se obtendrá el producto de sustitución.
COMPETENCIA ENTRE LAS REACCIONES
SN1 Y E1
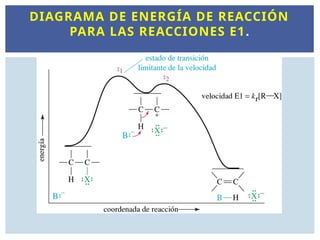
REACCIONES DE ELIMINACIÓNDE PRIMER
ORDEN E1
La etapa lenta es la formación del carbocatión y la velocidad de reacción
depende únicamente de la [RX ]. v = k[RX]
La reacción es de orden 1 por RX E1 (Eliminación de orden 1 o
unimolecular).
C
H
C
C
H
C
H
C C
C
X
C
B
C
X
-
Carbocatión
G
+ B-
+ X
+ BH + X
Coordenada de reacción
, B
70.
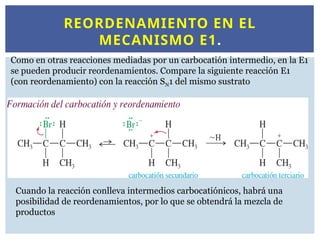
REORDENAMIENTO EN EL
MECANISMOE1.
Como en otras reacciones mediadas por un carbocatión intermedio, en la E1
se pueden producir reordenamientos. Compare la siguiente reacción E1
(con reordenamiento) con la reacción SN1 del mismo sustrato
Cuando la reacción conlleva intermedios carbocatiónicos, habrá una
posibilidad de reordenamientos, por lo que se obtendrá la mezcla de
productos
71.
Naturaleza del sustrato:La etapa limitante es la formación del
carbocatión, los más estables estarán estabilizados por efectos
inductivos y resonancia . Todo derivado que conduzca a un
carbocatión estable reaccionara según un mecanismo E1.
CH
C
B
B
CH3 C Br
CH3
CH3
CH3 CH2
CH3
H
C CH2
CH3
CH3
CH CH3
Cl
CH CH2
H
CH2
etapa
lenta
etapa
+
rapida
+ BH + Br-
+
+ BH + Cl-
FACTORES DETERMINANTES DE UNA E1
72.
Naturaleza del gruposaliente:
Cuanto más débil sea el enlace C-X, más fácil es la formación
del carbocatión (X debe ser atractor de electrones).
X debe ser un buen grupo saliente
Los iones de gran tamaño son los mejores grupos salientes.
Naturaleza de la base:
La base debe ser una base débil
< < < <
ROH, H2O
NATURALEZA DEL GRUPO SALIENTE Y LA
BASE EN LA ELIMINACIÓN E1
73.
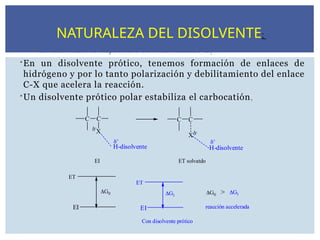
El disolventees importante en el mecanismo E1
En un disolvente prótico, tenemos formación de enlaces de
hidrógeno y por lo tanto polarización y debilitamiento del enlace
C-X que acelera la reacción.
Un disolvente prótico polar estabiliza el carbocatión,
C C
X
-
+
H-disolvente
C C
X
EI ET solvatdo
EI EI
G0
Con disolvente prótico
reacción accelerada
H-disolvente
ET
ET
G1
G0 G1
>
-
+
NATURALEZA DEL DISOLVENTE:
74.
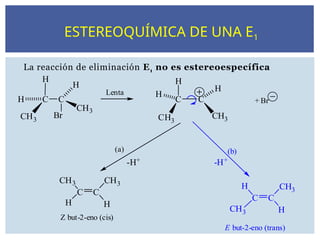
La reacción deeliminación E1 no es estereoespecífica
-H+ -H+
H C C
H
CH3
Br
H
CH3
C C
H
CH3
H
CH3
H
Lenta
+ Br
(a) (b)
C C
H
CH3
CH3
H
E but-2-eno (trans)
C C
CH3
H
CH3
H
Z but-2-eno (cis)
ESTEREOQUÍMICA DE UNA E1
75.
COMPETICIÓN SN1 –E1
a) Si el disolvente es polar y el nucleófilo es débilmente básico la
sustitución SN1 es favorita;
b) A bajas temperaturas la sustitución SN1 es favorita;
c) A altas temperaturas la eliminación E1 es favorita.
76.
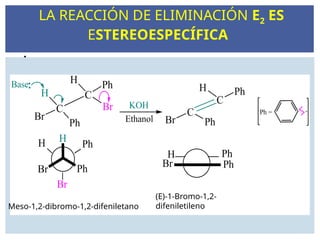
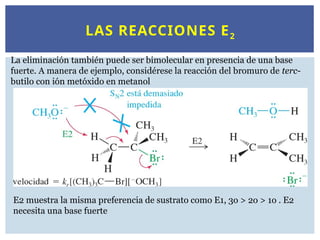
LAS REACCIONES E2
Laeliminación también puede ser bimolecular en presencia de una base
fuerte. A manera de ejemplo, considérese la reacción del bromuro de terc-
butilo con ión metóxido en metanol
E2 muestra la misma preferencia de sustrato como E1, 3o > 2o > 1o . E2
necesita una base fuerte
77.
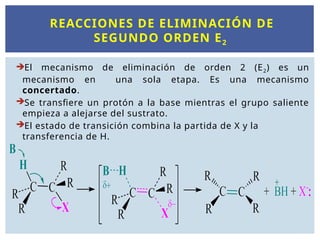
REACCIONES DE ELIMINACIÓNDE
SEGUNDO ORDEN E2
El mecanismo de eliminación de orden 2 (E2) es un
mecanismo en una sola etapa. Es una mecanismo
concertado.
Se transfiere un protón a la base mientras el grupo saliente
empieza a alejarse del sustrato.
El estado de transición combina la partida de X y la
transferencia de H.
78.
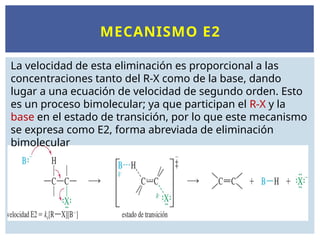
MECANISMO E2
La velocidadde esta eliminación es proporcional a las
concentraciones tanto del R-X como de la base, dando
lugar a una ecuación de velocidad de segundo orden. Esto
es un proceso bimolecular; ya que participan el R-X y la
base en el estado de transición, por lo que este mecanismo
se expresa como E2, forma abreviada de eliminación
bimolecular
79.
REACCIONES DE ELIMINACIÓNDE
SEGUNDO ORDEN E2
La velocidad es función de RX y de B-
v = k[RX][B-
]
La reacción es del orden 2 por RX et B-
E2 (eliminación de
segundo orden o bimolecular).
EF
EI
G
G°
ET
80.
Estados detransición concentrados de la reacción
E2. Los orbitales del átomo de hidrógeno y el haluro
deben estar alineados para que puedan comenzar a
formar un enlace pi en el estado de transición.
El hidrógeno que se va a abstraer debería ser anti-
coplanar al grupo saliente para minimizar cualquier
impedimento estérico entre la base y el grupo
saliente. Una orientación sin-coplanar entre el
hidrógeno y el grupo saliente creará una interacción
repulsiva y aumentará la energía del estado de
transición.
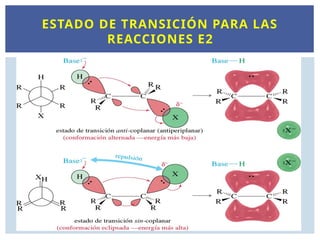
ESTADO DE TRANSICIÓN PARA LAS
REACCIONES E2
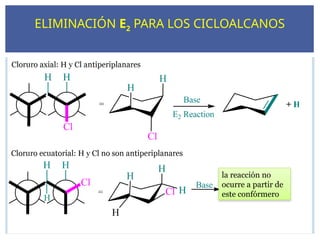
La reacción deeliminación E2 es estereoespecífica
H y X deben ser antiperiplanares
para tener una eliminación anti.
El solapamiento de los orbitales π en el ET requiere una
geometría antiperiplanar.
H
Br
H
Br
H
H
H H
Antiperiplana
r
Estado de transición anti
Producto: Alqueno
ESTEREOQUÍMICA DE LA E2
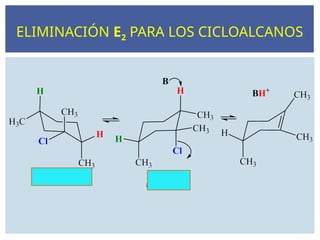
ELIMINACIÓN E2 PARALOS CICLOALCANOS
la reacción no
ocurre a partir de
este confórmero
88.
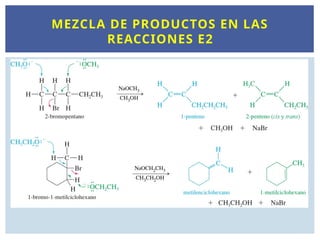
Las reaccionesE2 requieren la abstracción de un
protón de un átomo de carbono próximo al
carbono que lleva el halógeno. Si hay dos o más
posibilidades, se obtienen mezclas de productos.
Los ejemplos siguientes muestran cómo la
abstracción de protones diferentes puede dar
lugar a productos diferentes.
En general, el alqueno más sustituido será el
producto principal de la reacción
MEZCLA DE PRODUCTOS EN LAS
REACCIONES E2
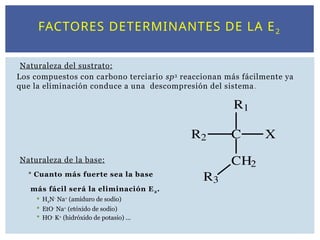
FACTORES DETERMINANTES DELA E2
Naturaleza del sustrato:
Los compuestos con carbono terciario sp3
reaccionan más fácilmente ya
que la eliminación conduce a una descompresión del sistema.
Naturaleza de la base:
Cuanto más fuerte sea la base
más fácil será la eliminación E2.
H2N-
Na+
(amiduro de sodio)
EtO-
Na+
(etóxido de sodio)
HO-
K+
(hidróxido de potasio) ...
C
CH2
X
R2
R3
R1
91.
En un disolventeprótico la fuerza de la base disminuye, por
formación de enlaces de H2, por lo que la E2 está desfavorecida.
Al contrario, en un disolvente aprótico y polar (DMSO), la fuerza
de la base se refuerza y la reacción E2 se acelera.
B H - so lvan t
B H
C C
+
+
-
X
-
d isp ersio n d e la ch arge
ET
B-
M+
O S
CH3
CH3
NATURALEZA DEL DISOLVENTE
92.
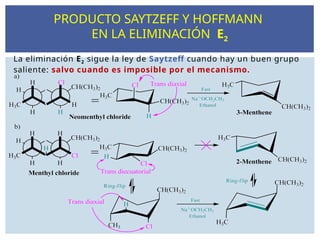
La reacción deeliminación E2 sigue la ley de Saytzeff cuando
hay un buen grupo saliente, y da como producto un alqueno más
sustituido y sigue la ley de Hoffmann cuando da como producto
un alqueno menos sustituido.
CH
B B
CH3
H
CH CH2
Br
H
1 2
2 productos
1 2
CH3 - CH = CH- CH3
Isómero más sustituido
CH3 - CH2 - CH= CH2
Isómero menos sustituido
Regla de Saytzeff Regla de Hoffmann
el más estable (mayoritario) el menos estable(minoritario)
PRODUCTO SAYTZEFF Y HOFFMANN
EN LA ELIMINACIÓN E2
93.
La eliminación E2sigue la ley de Saytzeff cuando hay un buen grupo
saliente: salvo cuando es imposible por el mecanismo.
PRODUCTO SAYTZEFF Y HOFFMANN
EN LA ELIMINACIÓN E2
COMPETICIÓN SN2 –E2
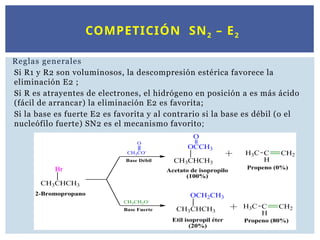
Reglas generales
Si R1 y R2 son voluminosos, la descompresión estérica favorece la
eliminación E2 ;
Si R es atrayentes de electrones, el hidrógeno en posición a es más ácido
(fácil de arrancar) la eliminación E2 es favorita;
Si la base es fuerte E2 es favorita y al contrario si la base es débil (o el
nucleófilo fuerte) SN2 es el mecanismo favorito;
98.
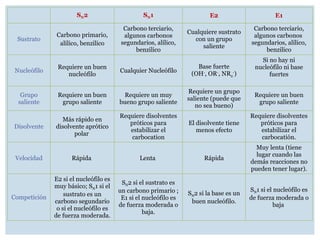
SN2 SN1 E2E1
Sustrato
Carbono primario,
alilico, benzilico
Carbono terciario,
algunos carbonos
segundarios, alílico,
benzilico
Cualquiere sustrato
con un grupo
saliente
Carbono terciario,
algunos carbonos
segundarios, alílico,
benzilico
Nucleófilo
Requiere un buen
nucleófilo
Cualquier Nucleófilo
Base fuerte
(OH-
, OR-
, NR2
-
)
Si no hay ni
nucleófilo ni base
fuertes
Grupo
saliente
Requiere un buen
grupo saliente
Requiere un muy
bueno grupo saliente
Requiere un grupo
saliente (puede que
no sea bueno)
Requiere un buen
grupo saliente
Disolvente
Más rápido en
disolvente aprótico
polar
Requiere disolventes
próticos para
estabilizar el
carbocation
El disolvente tiene
menos efecto
Requiere disolventes
próticos para
estabilizar el
carbocatión.
Velocidad Rápida Lenta Rápida
Muy lenta (tiene
lugar cuando las
demás reacciones no
pueden tener lugar).
Competición
E2 si el nucleófilo es
muy básico; SN1 si el
sustrato es un
carbono segundario
o si el nucleófilo es
de fuerza moderada.
SN2 si el sustrato es
un carbono primario ;
E1 si el nucleófilo es
de fuerza moderada o
baja.
SN2 si la base es un
buen nucleófilo.
SN1 si el nucleófilo es
de fuerza moderada o
baja






![La reacción es del orden 2 (1 respecto a RX y 1
respecto Nu-
) SN2 (sustitución nucleofílica de
segundo orden o bimolecular). Esto se debe a que
en el estado de transición interactúan el
halogenuro y el nucleófilo, por lo que se dice que
la velocidad es función de RX y de Nu-
v = k[RX][Nu-
]
En consecuencia esta reacción tiene una cinética
de segundo orden.
CINETICA DE LA REACCION SN2](https://image.slidesharecdn.com/halurosdealquilo-250603212108-b7cac7f0/85/Haluros-de-alquilo-14-320.jpg)





![La velocidad de reacción depende sólo de la etapa
lenta y por lo tanto sólo de la concentración del
sustrato RX y no de la del nucleófilo Nu-
.
v = k[RX]
La ecuación de velocidad es de primer orden: primer
orden respecto a la concentración del haluro de alquilo
y de orden cero respecto a la concentración del
nucleófilo.
CINETICA DE LA REACCION SN1](https://image.slidesharecdn.com/halurosdealquilo-250603212108-b7cac7f0/85/Haluros-de-alquilo-32-320.jpg)







![COMPARACIÓN SN1 Y SN2
SN1 SN2
Factores que influyen
Nucleófilo
Son apropiados los nucleófilos
débiles
Se necesitan nucleófilos
fuertes
Sustrato (RX) 3º > 2º CH3X > 1º > 2º
Disolvente
Se necesitan disolventes
ionizantes buenos
Amplia variedad de
disolventes
Grupo Saliente Ha de ser bueno Ha de ser bueno
Características
Cinéticas Primer orden kr[RX]
Segundo orden, kr[RX]
[Nu-
]
Estereoquímica
Mezcla de inversión y
retención
Inversión completa
Transposiciones Común Imposible](https://image.slidesharecdn.com/halurosdealquilo-250603212108-b7cac7f0/85/Haluros-de-alquilo-60-320.jpg)




![REACCIONES DE ELIMINACIÓN DE PRIMER
ORDEN E1
La etapa lenta es la formación del carbocatión y la velocidad de reacción
depende únicamente de la [RX ]. v = k[RX]
La reacción es de orden 1 por RX E1 (Eliminación de orden 1 o
unimolecular).
C
H
C
C
H
C
H
C C
C
X
C
B
C
X
-
Carbocatión
G
+ B-
+ X
+ BH + X
Coordenada de reacción
, B](https://image.slidesharecdn.com/halurosdealquilo-250603212108-b7cac7f0/85/Haluros-de-alquilo-69-320.jpg)



![REACCIONES DE ELIMINACIÓN DE
SEGUNDO ORDEN E2
La velocidad es función de RX y de B-
v = k[RX][B-
]
La reacción es del orden 2 por RX et B-
E2 (eliminación de
segundo orden o bimolecular).
EF
EI
G
G°
ET](https://image.slidesharecdn.com/halurosdealquilo-250603212108-b7cac7f0/85/Haluros-de-alquilo-79-320.jpg)