Introducción
• La termodinámicaes la parte de la Física que estudia los intercambios de
calor y trabajo que acompañan a los procesos fisicoquímicos- Si estos procesos
son reacciones químicas, hablamos de termoquímica.
Conceptos Previos
SISTEMA TERMODINÁMICO
•Los sistemas se clasifican en función de su capacidad de intercambio de
materia y/o energía (calor o trabajo):
• Abiertos: intercambian materia y energía con el entorno
• Cerrados: no intercambian materia pero sí energía (calor y trabajo)
• Aislados: no intercambian ni materia ni energía
• Adiabático: no intercambia materia ni energía en forma de calor; solo
intercambia energía en forma de trabajo.
5.
Conceptos Previos
SISTEMA TERMODINÁMICO
•Las reacciones químicas son sistemas termodinámicos
(reactivos + productos).
• Reacciones exotérmicas: ceden calor al entorno (-). La
energía de los reactivos es mayor que la energía de los
productos.
∆𝐸 = 𝐸𝑅 − 𝐸𝑃 ; ∆𝐸 < 0
• Reacciones enfotérmicas: toman calor al entorno (+).
La energía de los reactivos es menor que la energía de
los productos.
∆𝐸 = 𝐸𝑅 − 𝐸𝑃 ; ∆𝐸 > 0
6.
Conceptos Previos
VARIABLES TERMODINÁMICAS
•Los sistemas termodinámicos están definidos por diferentes variables
termodinámicas. Estas pueden ser:
• Variables de estado: magnitudes que solo dependen del estado inicial y
final del sistema independientemente del proceso seguido.
• Variables de transferencia: magnitudes cuyo valor sí que depende del
proceso seguido desde el estado inicial al estado final.
• Son:
Variables de estado Variables de transferencia
Presión (P) Volumen (V) Temperatura (T) Calor (Q) Trabajo (W)
7.
Conceptos Previos
PROCESO TERMODINÁMICO
•Los sistemas van a evolucionar desde un estado inicial a un estado final
mediante un proceso termodinámico:
• Los procesos termodinámicos se pueden llevar a cabo en diferentes condiciones en
función de qué magnitud permanezca invariable (constante). Son:
• Isobárico: la presión no varía (permanece constante).
• Isocórico: el volumen no varía
• Isotérmico: la temperatura no varía
8.
Calor
• Se denominacalor (Q) a la energía en tránsito que pasa de un cuerpo a otro cuando estos
están a distinta temperatura.
• Sus unidades serán las establecidas para la energía (J), aunque a menudo se mida en
calorías (cal) o en kilocalorías (kcal)
1 J = 0,24 cal 1 cal = 4,18 J
9.
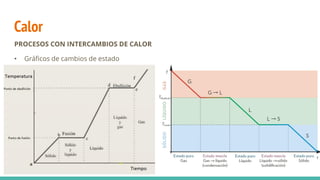
Calor
PROCESOS CON INTERCAMBIOSDE CALOR
• El intercambio de calor puede darse en 2 circunstancias:
• Cuando la temperatura se modifica: depende de la masa de sustancia, del incremento
de temperatura y de su calor específico*
* Capacidad calorífica (C) (C=m·ce)
*El calor específico, ce, indica la cantidad de calor necesaria para elevar la temperatura
por unidad de masa. Se expresa en cal/g·ºC o J/kg·K
𝑄 = 𝐶 · ∆𝑇 = 𝑚 · 𝑐𝑒 · ∆𝑇
10.
Calor
PROCESOS CON INTERCAMBIOSDE CALOR
• Cuando la temperatura no se modifica: se trata del calor que se aporta o se retira de un
sistema cuando se produce un cambio de estado.
*El calor latente, L, indica la cantidad de calor necesaria por unidad de masa para
producir un cambio de estado sin elevar la temperatura. Se expresa en cal/g o J/kg.
Puede ser
• Calor latente de fusión (Lf)
• Calor latente de vaporización (Lv)
𝑄 = 𝑚 · 𝐿
𝑆 → 𝐿
𝐿 → 𝑆
𝐿 → 𝐺
𝐺 → 𝐿
Calor
EJERCICIOS:
1. Calcula lacantidad de calor que hay que suministrar a un sistema formado por 80 g
de aluminio ( 𝒄𝒆 = 𝟗𝟏𝟎 𝑱/𝒌𝒈 · 𝑲 ) que se encuentra a 20ºC para que alcance la
temperatura de 80ºC.
Datos: Tf = 660ºC; Te = 2470ºC
13.
Calor
EJERCICIOS:
2. Calcula lacantidad de calor que hay que suministrar a 1 mol de agua en fase líquida
para que se transforme en vapor a 200ºC.
Datos: H: 1u; O: 16 u; 𝐿𝑣 = 2,2 · 106
𝐽/𝑘𝑔 ; 𝑐𝑒 = 2010 𝐽/𝑘𝑔 · 𝐾
14.
Calor
EJERCICIOS:
3. ¿Qué cantidadde calor hay que suministrar a 2 moles de agua a 25ºC para que
alcancen la temperatura de 110ºC?.
Datos: H: 1u; O: 16 u; 𝐿𝑣 = 2,2 · 106
𝐽/𝑘𝑔 ; 𝑐𝑒,𝑙𝑖𝑞𝑢𝑖𝑑𝑜 = 4180 𝐽/𝑘𝑔 · 𝐾 𝑐𝑒,𝑣𝑎𝑝𝑜𝑟 = 2010 𝐽/𝑘𝑔 · 𝐾
15.
Trabajo
• Se denominatrabajo (W) a la energía transferida a un sistema cuando sobre él actúa una
fuerza.
• El trabajo se mide en Julios (J) al ser una energía.
• Está referido a las expansiones y compresiones de los gases (cuando hay variación en el
volumen en recipientes cerrados con émbolo móvil).
Trabajo
• El trabajopuede calcularse mediante gráfico P-V
Proceso Isóbaro Proceso Isócoro Proceso Isotermo
𝑃 = 𝑐𝑡𝑒 𝑉 = 𝑐𝑡𝑒 𝑇 = 𝑐𝑡𝑒
𝑊 = −𝑃 · ∆𝑉
𝑊 = −𝑃 · (𝑉2 − 𝑉1)
𝑊 = 0 𝑊 = −𝑛𝑅𝑇 · 𝑙𝑛
𝑉2
𝑉1
18.
Trabajo
• ¿De dóndeviene la ecuación del W en el proceso isotermo?
𝑑𝑊 = −𝑃 · 𝑑𝑉
𝑑𝑊 = −
𝑛𝑅𝑇
𝑉
· 𝑑𝑉
𝑑𝑊 = −𝑛𝑅𝑇 ·
1
𝑉
𝑑𝑉
න
1
2
𝑑𝑊 = −𝑛𝑅𝑇 න
1
2
𝑑𝑉
𝑉
𝑊1→2 = −𝑛𝑅𝑇 · 𝑙𝑛𝑉2 − 𝑙𝑛𝑉1
𝑾𝟏→𝟐 = −𝒏𝑹𝑻 · 𝐥𝐧
𝑽𝟐
𝑽𝟏
19.
Trabajo
EJERCICIOS:
4. Observando lagráfica, calcula el trabajo de
expansión que realizan 250 g de Cl2 a 300 K a
través del camino A, del B y del C en su trayecto
desde 1 hasta 2.
a) ¿Se trata de un trabajo de expansión o de
compresión?
b) Tras finalizar el ejercicio, responde: ¿es W
una función de estado?
Datos: Cl: 35,5u; R: 0,082 atm·L/mol·K
20.
Trabajo
EJERCICIOS:
5. Calcula eltrabajo de expansión que experimenta un sistema formado por 7 g de
nitrógeno gaseoso que se hayan inicialmente a 1 atm de presión y una temperatura de
27ºC, cuando sigue estos procesos:
1º) Expansión a presión constante hasta duplicar su volumen, seguida de una
transformación a volumen constante hasta reducir su presión a la mitad.
2º) Transformación a volumen constante hasta reducir su presión a la mitad,
seguida de expansión a presión constante hasta duplicar su volumen
3º) Expansión isotérmica hasta que la presión se reduce a la mitad.
Datos: N: 14 u; R: 0,082 atm·L/mol·K
21.
1.Primer principio dela termodinámica
• La ley de conservación de la energía dice que la energía no se crea ni se
destruye, que simplemente se transforma y que, por tanto, permanece
constante.
• En termodinámica, los sistemas están formados por infinidad de partículas (EC,
EP, movimientos de rotación, vibración, etc.) A la suma de esas energías se le
denomina Energía Interna (U).
• Es una función de estado (depende de P, V y T) y se mide en J.
• No se puede medir su valor, pero sí su variación.
22.
1.Primer principio dela termodinámica
“Cuando un sistema (proceso químico” sufre una transformación termodinámica,
la variación de energía interna (∆𝑈) que experimenta es igual a la suma del calor
(desprendido o absorbido) más el trabajo (realizado por el sistema o sobre el
sistema) que el sistema intercambia con el entorno”.
∆𝑈 = 𝑄 + 𝑊
23.
1.Primer principio dela termodinámica
• Se debe establecer un criterio de signo. Escogeremos el criterio de signos
IUPAC.
Q>0 Q<0
W>0 W<0
24.
1.Primer principio dela termodinámica
TRUQUI: Todo lo que entra al sistema es positivo y todo lo que sale es negativo
Q>0 Q<0
W>0 W<0
CALOR (Q) TRABAJO (W)
Si el sistema toma calor, + Si es el entorno quien aplica un trabajo sobre el
sistema, disminuye de volumen, +
Si el sistema pierde calor, - Si el sistema realiza un trabajo sobre el entorno, es
decir, aumenta de volumen, -
1.Primer principio dela termodinámica
APLICACIONES DEL PRIMER PRINCIPIO DE LA TERMODINÁMICA
• Proceso Isotermo
• Muy complicados de llevar a cabo pues no se puede variar T
• ∆𝑼 = 𝟎 ya que solo depende de T. Todo el calor absorbido se emplea en un
trabajo de expansión.
∆𝑈 = 𝑄 + 𝑊
0 = 𝑄 + 𝑊
𝑸 = −𝑾
27.
1.Primer principio dela termodinámica
APLICACIONES DEL PRIMER PRINCIPIO DE LA TERMODINÁMICA
• Proceso isócoro
• El volumen permanece constante, luego no hay expansión ni compresión
→ 𝑾 = 𝟎.
• La variación de energía interna se consigue con la aplicación de calor
∆𝑈 = 𝑄 + 𝑊
∆𝑈 = 𝑄 + 0
∆𝑼 = 𝑸 ∆𝑼 = 𝑸𝐯
∆𝑈 = 𝑄 = 𝑚 · 𝑐𝑒 · ∆𝑇
∆𝑈 = 𝑄𝑣 = 𝑚 · 𝑐𝑣 · ∆𝑇
28.
1.Primer principio dela termodinámica
APLICACIONES DEL PRIMER PRINCIPIO DE LA TERMODINÁMICA
• Proceso Adiabático
• Al no haber intercambio de calor con el entorno, 𝑸 = 𝟎.
• La ∆𝑼 será:
∆𝑈 = 𝑄 + 𝑊
∆𝑈 = 0 + 𝑊
∆𝑼 = 𝑾
29.
1.Primer principio dela termodinámica
APLICACIONES DEL PRIMER PRINCIPIO DE LA TERMODINÁMICA
• Proceso isóbaro
• La presión es constante, luego, 𝑾 = −𝑷 · ∆𝑽.
• La ∆𝑼 será:
∆𝑈 = 𝑄 + 𝑊
∆𝑈 = 𝑄𝑃 + 𝑊
∆𝑼 = 𝑸𝑷 − 𝑷 · ∆𝑽
30.
1.Primer principio dela termodinámica
APLICACIONES DEL PRIMER PRINCIPIO DE LA TERMODINÁMICA
Isotermo Isócoro Adiabático Isóbaro
∆𝑈 0 𝑄𝑣 W 𝑄 + 𝑊
𝑄 -W 𝑚 · 𝑐𝑣 · ∆𝑇 0 𝑚 · 𝑐𝑒 · ∆𝑇
𝑊 -Q 0 ∆𝑈 −𝑃 · ∆𝑉
31.
1.Primer principio dela termodinámica
EJERCICIOS:
6. Un sistema realiza un trabajo de 150 J sobre el entorno y absorbe 80 J de calor. Halla
la variación de energía interna del sistema.
32.
1.Primer principio dela termodinámica
EJERCICIOS:
7. Al quemarse la gasolina en un cilindro del motor de un coche, se liberan 120 kJ. Si el
trabajo realizado por los gases producidos en la combustión es de 50 kJ, calcule la
variación de energía interna del sistema.
33.
1.Primer principio dela termodinámica
EJERCICIOS:
8. Calcule la variación de energía interna cuando 1 mol de agua a 30ºC aumenta su
temperatura hasta 70ºC manteniendo constante la presión.
Datos: 𝑐𝑒 = 4180 𝐽/𝑘𝑔 · 𝐾
34.
1.Primer principio dela termodinámica
EJERCICIOS:
9. Calcule la variación de energía interna que experimenta un sistema formado por 1
mol de agua líquida a 100ºC que se calienta hasta los 200 ºC. Se considera que la
presión es constante y de 1 atm y que el vapor de agua se comporta como gas ideal.
Datos: 𝑐𝑒,𝑣𝑎𝑝 = 2010 𝐽/𝑘𝑔 · 𝐾 𝐿𝑣 = 2,2 · 106
𝐽/𝑘𝑔 𝜌𝑎𝑔𝑢𝑎 = 1 𝑔/𝑚𝑙
35.
1.Primer principio dela termodinámica
EJERCICIOS:
10. ¿Qué variación de energía interna se produce al transformarse 100 g de agua a 25ºC
en vapor de agua a 100ºC a la presión constante de 101325 Pa?
Datos: 𝑐𝑒,𝑙𝑖𝑞𝑢 = 4180 𝐽/𝑘𝑔 · 𝐾 𝐿𝑣 = 2,2 · 106
𝐽/𝑘𝑔 𝜌𝑎𝑔𝑢𝑎 = 1000 𝑘𝑔/𝑚3 𝑅 = 8,31 𝐽/𝑚𝑜𝑙 · 𝐾
36.
2.Entalpía
• La mayoríade procesos ocurren a P=cte:
• La entalpía (∆𝐻) de un sistema representa el calor que intercambia con el entorno
cuando el proceso se realiza a P=cte.
• Se trata de una función de estado al ser combinación de funciones de estado (U, P y V). Se
mide en J.
∆𝑈 = 𝑄𝑃 − 𝑃∆𝑉 → 𝑑𝑒𝑠𝑝𝑒𝑗𝑎𝑛𝑑𝑜 𝑄𝑃
𝑄𝑃 = ∆𝑈 + 𝑃∆𝑉 = U2 − U1 + P V2 − V1
= U2 + PV2 − U1 + PV1 = H2 − H1 = ∆𝐻
37.
2.Entalpía
• En sólidosy líquidos donde ∆𝑉≈0 (no hay trabajo de expansión)
• Si intervienen gases ideales:
𝑄𝑃 = ∆𝐻 = ∆𝑈 + 𝑃∆𝑉 = ∆𝑈
𝑄𝑃 = 𝑄𝑉
∆𝐻 = ∆𝑈 + 𝑃∆𝑉 ; ∆𝐻 = ∆𝑈 + ∆ 𝑛𝑅𝑇
∆𝐻 = ∆𝑈 + ∆𝑛𝑅𝑇 ; 𝑄𝑃 = 𝑄𝑣 + ∆ 𝑛𝑅𝑇
38.
2.Entalpía
• Cuando haymás moles en productos que en reactivos:
• Cuando hay más moles en productos que en reactivos:
𝑄𝑃 > 𝑄𝑣 𝑝𝑜𝑟𝑞𝑢𝑒 QP = Qv + ∆𝑛𝑅𝑇
¿Cómo es el calor
que se libera a
P=cte y V=cte?
𝑄𝑃 < 𝑄𝑣 𝑝𝑜𝑟𝑞𝑢𝑒 QP = Qv + ∆𝑛𝑅𝑇
+
-
39.
2.Entalpía
• Una ecuacióntermoquímica incluye:
• Reacción química ajustada con estados de agregación
• P y T
• Cantidad de calor intercambiada con el entorno (∆𝐻)
• Si ∆𝐻 incorpora º → condiciones estándar (1 atm y 25ºC)
• Como ∆𝐻 = Hproductos − Hreactivos
∆𝐻 < 0 → 𝑒𝑥𝑜𝑡é𝑟𝑚𝑖𝑐𝑎
∆𝐻 > 0 → 𝑒𝑛𝑑𝑜𝑡é𝑟𝑚𝑖𝑐𝑎
40.
2.Entalpía
• La representacióngráfica del contenido energético de reactivos y productos y la variación
entálpica del proceso se denomina diagrama entálpico.
2.Entalpía
EJERCICIOS:
11. La variaciónde entalpía del proceso en el que se quema 1 mol de gas butano para
dar dióxido de carbono y agua líquida es de -2878 kJ/mol. Determina el calor que se
desprende si el proceso tiene lugar a volumen constante y a 25ºC.
43.
2.Entalpía
EJERCICIOS:
12. Se queman25 g de octano líquido (C8H18) a volumen constante a 25ºC
desprendiéndose 1200 kJ. ¿Cuál será ∆𝑼 y ∆𝑯 en la combustión de 3 moles de octano a
25ºC?.
Datos: C: 12 u; H: 1 u; R=8,31 J/mol·K
44.
2.Entalpía
EJERCICIOS:
13. A continuaciónse muestra el diagrama entálpico de una reacción química. Escribe
su ecuación termoquímica. ¿Se trata de una reacción endotérmica o exotérmica?
45.
3.Determinación de laEntalpía
EXPERIMENTALMENTE
• Se pueden determinar entalpías en reacciones de neutralización o
disolución
• Se utiliza un calorímetro.
• Se miden las temperaturas al inicio y al final del sistema.
• Utilizando un equivalente en agua (magua que absorbe el mismo
calor que calentando)
𝑄𝑑𝑒𝑠𝑝𝑟𝑒𝑛𝑑𝑖𝑑𝑜 = 𝑄𝑎𝑏𝑠𝑜𝑟𝑏𝑖𝑑𝑜 𝑝𝑜𝑟 𝑑𝑖𝑠𝑜𝑙𝑢𝑐𝑖𝑜𝑛𝑒𝑠 + 𝑄𝑎𝑏𝑠𝑜𝑟𝑏𝑖𝑑𝑜 𝑝𝑜𝑟 𝑐𝑎𝑙𝑜𝑟í𝑚𝑒𝑡𝑟𝑜
= 𝑚 · 𝑐𝑒𝑑𝑖𝑠𝑜𝑙𝑢𝑐𝑖𝑜𝑛𝑒𝑠 · (𝑇𝑓 − 𝑇𝑖) + 𝑚𝑒𝑞𝑎𝑔𝑢𝑎
· 𝑐𝑒𝑎𝑔𝑢𝑎 · (𝑇𝑓 − 𝑇𝑖)
46.
3.Determinación de laEntalpía
EJERCICIOS:
14. En un calorímetro cuyo equivalente en agua es 25 g se introducen 100 ml de
disolución de HCl 0,5 M y 100 ml de NaOH 0,5 M. La temperatura inicial del sistema
(disoluciones + calorímetro) es de 18ºC y la temperatura final de 22ºC. Determine la
entalpía de neutralización entre el HCl y el NaOH
Datos: 𝑐𝑒,𝑙𝑖𝑞𝑢 = 4180 𝐽/𝑘𝑔 · 𝐾 𝜌𝑎𝑔𝑢𝑎 = 1 𝑔/𝑚𝑙
47.
3.Determinación de laEntalpía
EJERCICIOS:
15. En un calorímetro cuyo equivalente en agua es 25 g se introducen 100 ml de agua y
se comprueba que la temperatura del sistema es de 18ºC. Se añaden 2 g de lentejas de
NaOH que, al disolverse, hacen que la temperatura del sistema llegue a 22ºC.
Determine la variación de entalpía del proceso de disolución del NaOH.
Datos: 𝑐𝑒,𝑙𝑖𝑞𝑢 = 4180 𝐽/𝑘𝑔 · 𝐾 𝜌𝑎𝑔𝑢𝑎 = 1 𝑔/𝑚𝑙
48.
3.Determinación de laEntalpía
LEY DE HESS
• Debido a que ∆𝐻 es una función de estado, el cálculo de esta puede realizarse por
combinación de otros procesos conocidos sin necesidad de llevarlo a cabo.
• La Ley de Hess establece que la variación de entalpía de un proceso es igual a la suma de
las variaciones de entalpía de cada uno de los subprocesos que dan lugar al mismo
proceso global.
3.Determinación de laEntalpía
LEY DE HESS – IMPORTANTE
• La entalpía es una magnitud extensiva, es decir, depende de la cantidad de materia:
• Si se invierte una reacción química, cambia el signo de su ∆𝐻.
• Si una ecuación química se multiplica por un factor, ∆𝐻 también se multiplica por ese
factor.
𝐶 𝑠 +
1
2
𝑂2 𝑔 → 𝐶𝑂 𝑔 ∆𝐻 = −110,5 𝑘𝐽
𝐶𝑂 𝑔 → 𝐶 𝑠 +
1
2
𝑂2 𝑔 ∆𝐻 = +110,5 𝑘𝐽
2 𝐶 𝑠 + 𝑂2 𝑔 → 2 𝐶𝑂 𝑔 ∆𝐻 = 2 · −110,5 = −221 𝑘𝐽
51.
3.Determinación de laEntalpía
LEY DE HESS – PASOS
1. Escribir ecuación de interés sin coeficientes fraccionarios.
2. Se buscan las ecuaciones que permitan obtener el proceso deseado.
3. Se comprueba que las sustancias están en la posición correcta (reactivos o productos). Si
no es así, se invierte la reacción y el signo de ∆𝐻.
4. Se comprueban los coeficientes estequiométricos. Si no son los del proceso deseado, se
multiplican o dividen.
5. Suma de ecuaciones y suma de entalpías
6. Referir a 1 mol
52.
3.Determinación de laEntalpía
EJEMPLO
El peróxido de hidrógeno, conocido como agua oxigenada, se descompone fácilmente y se
transforma en agua y oxígeno. Calcula la entalpía del proceso en el cual se forma agua
oxigenada a partir del hidrógeno y el oxígeno.
2 𝐻2 𝑔 + 𝑂2 𝑔 → 2 𝐻2𝑂 𝑙 ∆𝐻 = −571 𝑘𝐽
𝐻2𝑂2 𝑙 → 𝐻2𝑂 𝑙 +
1
2
𝑂2 𝑔 ∆𝐻 = −98 𝑘𝐽
3.Determinación de laEntalpía
EJERCICIOS
16. Determina la entalpía del proceso en el cual se forma 1 mol de N2O5 (g) a partir de
los elementos que lo integran. Utiliza los siguientes datos.
A 𝑁2 𝑔 + 3 𝑂2 𝑔 + 𝐻2 𝑔 → 2 𝐻𝑁𝑂3 (𝑎𝑞) ∆𝐻º = −414,7 𝑘𝐽
B 𝑁2𝑂5 𝑔 + 𝐻2𝑂 𝑙 → 2 𝐻𝑁𝑂3 (𝑎𝑞) ∆𝐻º = −140,2 𝑘𝐽
C 2 𝐻2 𝑔 + 𝑂2 𝑔 → 2 𝐻2𝑂 𝑙 ∆𝐻º = −571,7 𝑘𝐽
55.
3.Determinación de laEntalpía
EJERCICIOS
17. Utiliza la ley de Hess y los siguientes datos para obtener la entalpía de la reacción de
formación del PCl5.
A 𝑃4 𝑠 + 6 𝐶𝑙2 𝑔 → 4 𝑃𝐶𝑙3 (𝑙) ∆𝐻º = −1270 𝑘𝐽
B 𝑃𝐶𝑙3 𝑙 + 𝐶𝑙2 (𝑔) → 𝑃𝐶𝑙5 (𝑠) ∆𝐻º = −137 𝑘𝐽
56.
3.Determinación de laEntalpía
EJERCICIOS
18. Calcula la entalpía de la reacción de formación del óxido de cinc a partir de los
siguientes datos.
A 𝐻2𝑆𝑂4 𝑎𝑞 + 𝑍𝑛𝑂 𝑠 → 𝑍𝑛𝑆𝑂4 𝑎𝑞 + 𝐻2𝑂 (𝑙) ∆𝐻º = −210 𝑘𝐽
B 𝐻2𝑆𝑂4 𝑎𝑞 + 𝑍𝑛 𝑠 → 𝑍𝑛𝑆𝑂4 𝑎𝑞 + 𝐻2 (𝑔) ∆𝐻º = −335 𝑘𝐽
C 2 𝐻2 𝑔 + 𝑂2 𝑔 → 2 𝐻2𝑂 𝑙 ∆𝐻º = −571 𝑘𝐽
57.
3.Determinación de laEntalpía
ENTALPÍA DE FORMACIÓN ESTÁNDAR
• El ∆𝑯º𝒇 de un compuesto es la variación de entalpía del proceso en el cual se forma 1 mol
de dicho compuesto a partir de sus elementos en su estado físico más estable.
• Se establece por convenio que ∆𝑯º𝒇 de los elementos en condiciones estándar (25ºC y 1
atm) (estado termodinámico más estable) es cero.
• Los ∆𝐻º𝑓 de las sustancias permiten calcular ∆𝐻º de las reacciones.
• Se requieren anexos de tablas de datos.
∆𝐻º = 𝑛 · ∆𝐻º𝑓 𝑝𝑟𝑜𝑑𝑢𝑐𝑡𝑜𝑠 − 𝑛 · ∆𝐻º𝑓(𝑟𝑒𝑎𝑐𝑡𝑖𝑣𝑜𝑠)
58.
3.Determinación de laEntalpía
EJERCICIOS
19. Calcula la entalpía estándar de la siguiente reacción utilizando las entalpías de
formación estándar tabuladas:
𝐶𝑂 𝑔 +
1
2
𝑂2 𝑔 → 𝐶𝑂2 𝑔
59.
3.Determinación de laEntalpía
EJERCICIOS
20. Para la fabricación industrial de ácido nítrico, la reacción de partida es la oxidación
del amoníaco:
Calcula ∆𝑯º a partir de las entalpías de formación estándar (∆𝑯º𝒇) tabuladas de las
sustancias implicadas.
4 𝑁𝐻3 𝑔 + 5 𝑂2 𝑔 → 6 𝐻2𝑂 𝑔 + 4 𝑁𝑂2 𝑔
60.
3.Determinación de laEntalpía
ENTALPÍA DE PROCESOS ESPECÍFICOS
• COMBUSTIÓN: es la variación de entalpía del proceso en el cual 1 mol de dicha sustancia
reacciona con O2 para dar CO2 (g) y H2O (l)
• HIDROGENACIÓN: es la variación de entalpía del proceso en el cual 1 mol de una sustancia
insaturada capta H2 para transformarse en la sustancia saturada correspondiente.
• Para este tipo de procesos, se escriben las ecuaciones químicas (termoquímicas) y se
aplica la ley de Hess.
61.
3.Determinación de laEntalpía
EJERCICIOS
21. Durante la combustión de 1 mol de átomos de azufre en condiciones estándar se
desprenden 296,8 kJ y durante la combustión de 1 mol de sulfuro de hidrógeno, 560 kJ.
Con estos datos, determina la variación de entalpía que se produce en el proceso:
2 𝐻2𝑆 𝑔 + 𝑆𝑂2 𝑔 → 2 𝐻2𝑂 𝑙 + 3 𝑆 𝑠
62.
3.Determinación de laEntalpía
EJERCICIOS
22. Calcula la entalpía estándar de formación del metano (CH4) a partir de las entalpías
estándar de combustión del C (s), H2 (g) y CH4 (g) cuyos valores son, respectivamente,
-393,5 kJ/mol, -285,9 kJ/mol y -890,4 kJ/mol.
63.
3.Determinación de laEntalpía
ENERGÍAS DE ENLACE
• La entalpía de enlace es la energía que se requiere para romper 1 mol de enlaces
establecidos entre dos átomos que se hallanen un compuesto en estado gaseoso.
• Los datos de las entalpías de enlace están tabulados.
∆𝐻º = 𝑛 · ∆𝐻º𝑓 𝑒𝑛𝑙𝑎𝑐𝑒𝑠 𝑟𝑜𝑡𝑜𝑠 − 𝑛 · ∆𝐻º𝑓(𝑒𝑛𝑙𝑎𝑐𝑒𝑠 𝑓𝑜𝑟𝑚𝑎𝑑𝑜𝑠)
64.
3.Determinación de laEntalpía
EJERCICIOS
23. Calcula la entalpía de hidrogenación del etileno para formar etano según la
reacción:
𝐶𝐻2 = 𝐶𝐻2 𝑔 + 𝐻2 𝑔 → 𝐶𝐻3 − 𝐶𝐻3 𝑔
65.
3.Determinación de laEntalpía
EJERCICIOS
24. A partir de las energías de enlace tabuladas, determinar la entalpía estándar de la
reacción:
𝐶𝐻4 𝑔 + 𝐶𝑙2 𝑔 → 𝐶𝐻3𝐶𝑙 𝑔 + 𝐻𝐶𝑙 𝑔
66.
3.Determinación de laEntalpía
EJERCICIOS
25. A 25ºC y 1 atm, la entalpía de formación del bromano es de 36,2 kJ/mol. Calcula la
entalpía de la reacción de disociación del HBr en sus átomos constituyentes sabiendo
que en las condiciones señaladas, las entalpías de disociación del H2 (g) y del Br2 (g) son
respectivamente, 435,6 kJ/mol y 193,28 kJ/mol.:
67.
4.Segundo principio dela termodinámica
• El primer principio de la termodinámica es una aplicación de la ley de conservación de la
energía, de modo que la energía del universo no varía.
• Sin embargo, aún cuando se conserva la energía del universo se dan transformaciones
espontáneas (sin ninguna acción externa) y no espontáneas (requieren de acción externa).
Hielo Agua liquida Rotura de objeto
Derribo de un edificio
Recipientes comunicantes
Evaporación del alcohol
68.
4.Segundo principio dela termodinámica
• El segundo principio de la termodinámica establece que en cualquier proceso natural, la
entropía de un sistema aislado tiende a aumentar. Es decir, los sistemas tienden a
evolucionar hacia un estado de mayor desorden o caos.
• La dirección de los procesos espontáneos: En cualquier proceso que ocurra
espontáneamente, la entropía total del sistema y su entorno aumenta. Este aumento
de entropía indica que los procesos naturales no pueden revertirse sin añadir trabajo
o energía externa. Por ejemplo, si dejas caer un vaso de cristal, se romperá de forma
espontánea, pero para volver a juntar los pedazos de cristal tendrías que aplicar
energía.
• La imposibilidad de una máquina térmica 100% eficiente: Este principio también se
relaciona con la imposibilidad de construir una máquina que convierta toda la
energía térmica en trabajo útil. Siempre habrá una parte de la energía que se disipa
como calor, aumentando la entropía del entorno.
69.
4.Segundo principio dela termodinámica
• ¿Qué tienen en común todos estos procesos espontáneos?
Aumento del desorden del universo
• Para predecir el grado de espontaneidad de los procesos hay que introducir una magnitud
nueva que pueda medir el universo.
ENTROPÍA (∆𝑆)
“Considerando al universo como un sistema aislado, a volumen constante se
producirán procesos espontáneos en los que la entropía aumenta.
∆𝑆𝑡𝑜𝑡𝑎𝑙 = (∆𝑆𝑠𝑖𝑠𝑡𝑒𝑚𝑎 + ∆𝑆𝑒𝑛𝑡𝑜𝑟𝑛𝑜) > 0
70.
4.Segundo principio dela termodinámica
ENTROPÍA
• Es una magnitud que mide el grado de desorden de un sistema. Se representa con la letra
S y se mide en J/K.
• Se trata de una función de estado, pues ∆𝑺 solo depende del estado inicial y final.
• En general, la S de un sistema aumenta con la T (por aumento de la Ec de las partículas).
• Sin embargo, la variación de entropía que un sistema experimenta no solo depende de la
temperatura sino también del calor que se aporta.
• Si un cuerpo tiene poca temperatura, pequeños aportes de calor pueden producir
una gran variación de entropía.
• Si un cuerpo tiene alta temperatura, el aporte de calor apenas repercutirá en el
aumento de su entropía
71.
4.Segundo principio dela termodinámica
ENTROPÍA
• La entropía se expresa como:
∆𝑆 =
∆𝑄
𝑇
• Siendo ∆𝑄 la cantidad de calor aportada de forma reversible a un sistema para producir un
cambio de temperatura absoluta (T).
72.
4.Segundo principio dela termodinámica
EJEMPLO:
26. Calcula la variación de entropía que experimenta 1 mol de agua a 100ºC cuando pasa
del estado gaseoso a la misma temperatura.
Datos: 𝐿𝑣 = 2,2 · 106
𝐽/𝑘𝑔
𝑄 = 𝑚 · 𝐿𝑣 = 18 · 10−3
𝑘𝑔 · 2,2 · 106 𝐽
𝑘𝑔
= 39600 𝐽
∆𝑆 =
𝑄
𝑇
=
39600 𝐽
373 𝐾
= 106,16 𝐽/𝐾
73.
4.Segundo principio dela termodinámica
27. Calcula e interpreta el signo de la variación de entropía de los siguientes procesos:
a) 100 g de agua a 0ºC se convierten en hielo y mantienen la temperatura.
Datos: 𝐿𝑓 = 0,33 · 106
𝐽/𝑘𝑔
b) 100 g de agua a 100ºC se convierten en vapor y mantienen la temperatura
Datos: 𝐿𝑣 = 2,2 · 106
𝐽/𝑘𝑔
74.
5.Tercer principio dela termodinámica
ENTROPÍA MOLAR ESTÁNDAR
• La entropía molar estándar (𝑆º𝑚) es el grado de desorden de un compuesto a 25ºC y 1 atm,
y se expresa en el SI en J/K·mol.
• Los valores se encuentran tabulados y se toma como referencia el 0 absoluto de
temperaturas y el tercer principio de la termodinámica.
“La entropía de un sólido cristalino perfectamente ordenado, en el cero absoluto de
temperatura, es cero (máximo orden).”
Así pues, todas las entropías van a ser positivas (𝑆>0).
75.
5.Tercer principio dela termodinámica
ENTROPÍA MOLAR ESTÁNDAR
• La entropía depende de:
• Estado de agregación
• Complejidad (nº enlaces)
• En reacciones químicas, la entropía molar se calcula a partir de las entropías de reactivo y
productos según la siguiente expresión:
∆𝑆º𝑚 = 𝑛𝑝𝑟𝑜𝑑𝑢𝑐𝑡𝑜𝑠 · ∆𝑆º𝑚 𝑝𝑟𝑜𝑑𝑢𝑐𝑡𝑜𝑠 − 𝑛𝑟𝑒𝑎𝑐𝑡𝑖𝑣𝑜𝑠 · ∆𝑆º𝑚(𝑟𝑒𝑎𝑐𝑡𝑖𝑣𝑜𝑠)
• La entropía aumentará si el número de partículas gaseosas es mayor en los productos que
en los reactivos y cuando una molécula se rompe en otras más pequeñas.
76.
5.Tercer principio dela termodinámica
28. Predice el signo de la variación de entropía de los siguientes procesos.
Posteriormente calcula el valor con los datos de la tabla y confirma la predicción.
𝐶2𝐻4 𝑔 + 𝐻2 𝑔 → 𝐶2𝐻6 (𝑔)
𝑆𝑂3 𝑔 + 𝐻2 𝑔 → 𝑆𝑂2 𝑔 + 𝐻2𝑂 (𝑔)
2 𝑂3 𝑔 → 3 𝑂2 (𝑔)
𝐶𝑙2 𝑔 + 𝐻2 𝑔 → 2 𝐻𝐶𝑙 (𝑔)
77.
6.Energía libre deGibbs y Espontaneidad
• Desde un punto de vista entálpico, serían procesos espontáneos los procesos exotérmicos
mientras que desde un punto de vista entrópico serían espontáneos los procesos acompañados
de un aumento del orden del sistema.
• Sin embargo, conocemos procesos endotérmicos como la evaporación del agua de los charcos y
procesos con disminución de la entropía como la cristalización de una sal.
• De este modo se ve que no es posible explicar la espontaneidad únicamente con ∆𝐻 e ∆𝑆. Surge
así una nueva magnitud:
ENERGÍA LIBRE DE GIBBS (∆𝐺)
78.
6.Energía libre deGibbs y Espontaneidad
• En un proceso cualquiera, la cantidad de calor que recibe el entorno coincide con la cantidad de calor que cede el sistema. Al ser el
entorno mucho más grande que el sistema, este intercambio de calor no modifica T (𝑇 = 𝑐𝑡𝑒).
∆𝑆𝑒𝑛𝑡𝑜𝑟𝑛𝑜 =
𝑄𝑒𝑛𝑡𝑜𝑟𝑛𝑜
𝑇
=
−𝑄𝑠𝑖𝑠𝑡𝑒𝑚𝑎
𝑇
𝑃=𝑐𝑡𝑒
∆𝑆𝑒𝑛𝑡𝑜𝑟𝑛𝑜 =
−∆𝐻𝑠𝑖𝑠𝑡𝑒𝑚𝑎
𝑇
• Aplicando el segundo principio de la termodinámica:
∆𝑆𝑢𝑛𝑖𝑣𝑒𝑟𝑠𝑜 = ∆𝑆𝑠𝑖𝑠𝑡𝑒𝑚𝑎 + ∆𝑆𝑒𝑛𝑡𝑜𝑟𝑛𝑜
∆𝑆𝑢𝑛𝑖𝑣𝑒𝑟𝑠𝑜 = ∆𝑆𝑠𝑖𝑠𝑡𝑒𝑚𝑎 +
−∆𝐻𝑠𝑖𝑠𝑡𝑒𝑚𝑎
𝑇
𝑇 · ∆𝑆𝑢𝑛𝑖𝑣𝑒𝑟𝑠𝑜 = 𝑇 · ∆𝑆𝑠𝑖𝑠𝑡𝑒𝑚𝑎 −∆𝐻𝑠𝑖𝑠𝑡𝑒𝑚𝑎
−𝑇 · ∆𝑆𝑢𝑛𝑖𝑣𝑒𝑟𝑠𝑜 = −𝑇 · ∆𝑆𝑠𝑖𝑠𝑡𝑒𝑚𝑎 +∆𝐻𝑠𝑖𝑠𝑡𝑒𝑚𝑎
∆𝐺 = −𝑇 · ∆𝑆 + ∆𝐻
∆𝑮 = ∆𝑯 − 𝑻 · ∆𝑺
79.
6.Energía libre deGibbs y Espontaneidad
• ∆𝐺º es una función de estado y cabe establecer:
∆𝐺º = 𝑛𝑝𝑟𝑜𝑑𝑢𝑐𝑡𝑜𝑠 · ∆𝐺º 𝑝𝑟𝑜𝑑𝑢𝑐𝑡𝑜𝑠 − 𝑛𝑟𝑒𝑎𝑐𝑡𝑖𝑣𝑜𝑠 · ∆𝐺º (𝑟𝑒𝑎𝑐𝑡𝑖𝑣𝑜𝑠)
• “La energía libre de formación estándar de un compuesto es la variación de energía libre del
proceso en el cual se forma 1 mol de ese compuesto a partir de sus elementos cuando estoss se
encuentran en condiciones estándar y en su estado termodinámico más estable. Para elementos
en condiciones estándar, ∆𝐺º𝑓 = 0.”
80.
6.Energía libre deGibbs y Espontaneidad
Se puede determinar la espontaneidad de un proceso a partir de su ∆𝐺:
• Si ∆𝑮 < 𝟎 → proceso espontáneo en el sentido descrito (irreversible).
• Si ∆𝐺 = 0 → el sistema está en equilibrio (reversible).
• Si ∆𝑮 > 𝟎 → proceso no espontáneo en el sentido descrito pero sí en el sentido inverso.
81.
6.Energía libre deGibbs y Espontaneidad
EVALUACIÓN DE LA ESPONTANEIDAD
Teniendo en cuenta la relación entre las magnitudes: ∆𝑮 = ∆𝑯 − 𝑻 · ∆𝑺
∆𝑯 ∆𝑺 ∆𝑮
∆𝐻 < 0 ∆𝑆 > 0
∆𝐺 < 0
Siempre espontáneo
∆𝐻 < 0 ∆𝑆 < 0
Depende de la temperatura:
• Si ∆𝐻 > 𝑇 · ∆𝑆 , ∆𝐺 < 0
(Temperaturas bajas = espontáneo)
• Si ∆𝐻 < 𝑇 · ∆𝑆 , ∆𝐺 > 0
(Temperaturas altas = no espontáneo)
∆𝐻 > 0 ∆𝑆 > 0
Depende de la temperatura:
• Si ∆𝐻 > 𝑇 · ∆𝑆 , ∆𝐺 > 0
(Temperaturas bajas = no espontáneo)
• Si ∆𝐻 < 𝑇 · ∆𝑆 , ∆𝐺 < 0
(Temperaturas altas = espontáneo)
∆𝐻 > 0 ∆𝑆 < 0 ∆𝐺 > 0
Siempre no espontáneo
82.
6.Energía libre deGibbs y Espontaneidad
EVALUACIÓN DE LA ESPONTANEIDAD
Cuando la espontaneidad de los procesos depende de la temperatura, es posible determinar el valor
de temperatura que diferencia un proceso espontáneo de uno no espontáneo. En el momento de
dicho cambio, ∆𝐺 = 0
∆𝑮 = ∆𝑯 − 𝑻 · ∆𝑺
𝟎 = ∆𝑯 − 𝑻 · ∆𝑺
∆𝑯 = 𝑻 · ∆𝑺
𝑻 =
∆𝑯
∆𝑺
CUIDADO
CON LAS
UNIDADES
83.
6.Energía libre deGibbs y Espontaneidad
29. Con los valores de las tablas, calcula ∆𝑯º y ∆𝑺º de los siguientes procesos. Utiliza
esos datos para calcular la ∆𝑮º a 25ºC de cada proceso. Calcula la ∆𝑮º con los valores
tabulados y comprueba la coincidencia:
𝐻𝐶𝑙 𝑔 + 𝑁𝐻3(𝑔) → 𝑁𝐻4𝐶𝑙 (𝑠)
𝐶6𝐻12 𝑔 + 9 𝑂2 𝑔 → 6 𝐶𝑂2 𝑔 + 6 𝐻2𝑂 (𝑙)
84.
6.Energía libre deGibbs y Espontaneidad
30. El clorato de potasio se descompone en cloruro de potasio y oxígeno. Las entalpías
de formación estándar del KCl y del KClO3 a 25ºC son, respectivamente, −𝟒𝟑𝟕 𝒌𝑱/𝒎𝒐𝒍 y
− 𝟑𝟗𝟖 𝒌𝑱/𝒎𝒐𝒍.
a) Calcular la variación de entalpía de la reacción e indica si es exotérmica o
endotérmica
b) Determinar cuál será el signo de la variación de entropía estándar de la
reacción
c) Justificar si la reacción será o no espontánea en condiciones estándar.
85.
6.Energía libre deGibbs y Espontaneidad
31. Calcular la temperatura de equilibrio para la reacción sin ajustar utilizando los
datos de las tablas:
𝑺𝑶𝟑 𝒈 → 𝑺𝑶𝟐 𝒈 + 𝑶𝟐(𝒈)
86.
6.Energía libre deGibbs y Espontaneidad
32. La reacción 𝑷𝑪𝒍𝟑 𝒈 + 𝑪𝒍𝟐 𝒈 → 𝑷𝑪𝒍𝟓 (𝒈), a 25ºC, presenta una ∆𝑮º = −𝟑𝟕, 𝟐 𝒌𝑱 y una
∆𝑯º = −𝟖𝟕, 𝟗 𝒌𝑱. Calcula:
a) La ∆𝑺º y la temperatura a partir de la cual el proceso no es espontáneo.
b) El intervalo de temperaturas en el que puede almacenarse el PCl5 (g) sin peligro
de que se descomponga.
87.
6.Energía libre deGibbs y Espontaneidad
33. Consultando las tablas de datos termodinámicos a 25ºC se tienen los siguientes valores:
Justifica si para dicha temperatura los siguientes enunciados son verdaderos o falsos:
a) La formación de NO a partir de nitrógeno y oxígeno en condiciones estándar es un
proceso endotérmico
b) El NO es una sustancia más estable que el NO2
c) La oxidación con oxígeno en condiciones estándar de NO a NO2 es exotérmica
d) La oxidación con oxígeno en condiciones estándar de NO a NO2 es espontánea
∆𝑯º𝒇 (kJ/mol) ∆𝑮º (kJ/mol)
NO (g) 90,25 86,57
NO2 (g) 33,18 51,30
88.
6.Energía libre deGibbs y Espontaneidad
34. A través de la fotosíntesis, los vegetales fabrican azúcares a partir del agua y el
dióxido de carbono del aire según la reacción simplificada:
𝟔 𝑪𝑶𝟐 𝒈 + 𝟔 𝑯𝟐𝑶 𝒈 → 𝑪𝟔𝑯𝟏𝟐𝑶𝟔 𝒔 + 𝟔 𝑶𝟐(𝒈)
a) Calcula la ∆𝑯º y ∆𝑺º de esta reacción. Razona, según criterios termodinámicos, por
qué es imposible que los vegetales puedan completar la fotosíntesis en condiciones
estándar a 25ºC sin una aportación de energía de una fuente externa.
b) La combustión regulada de los azúcares es la fuente de energía más importante de
los seres vivos. Calcula la ∆𝑯 correspondiente a la combustión de 25g de glucosa en
condiciones estándar a 25ºC y razona si la combustión de glucosa será un proceso
espontáneo o no desde un punto de vista termodinámico.