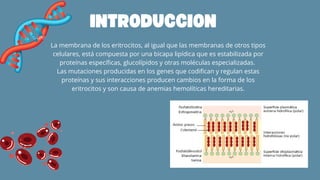
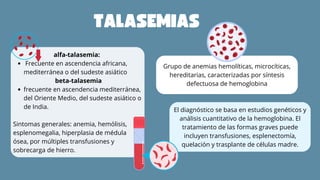
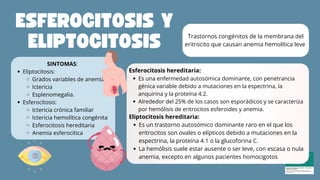
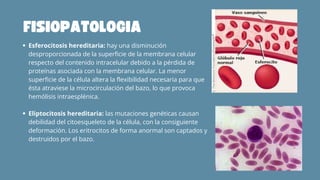
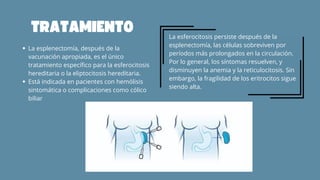
El documento aborda las anemias hemolíticas hereditarias causadas por defectos en la membrana de los eritrocitos, destacando tipos como la esferocitosis y eliptocitosis, así como su etiología y fisiopatología. Detalla los métodos de diagnóstico, incluidos análisis de sangre y pruebas genéticas, y los tratamientos que van desde esplenectomías hasta transfusiones sanguíneas. También se discuten las talasemias y su tratamiento, así como la importancia de la historia familiar en el diagnóstico.