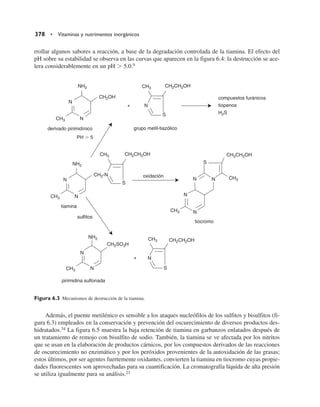
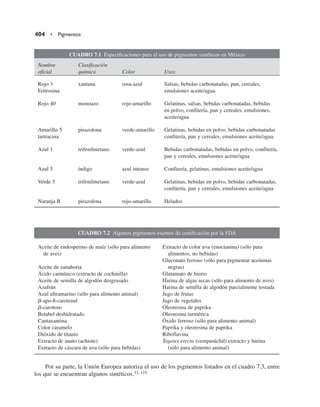
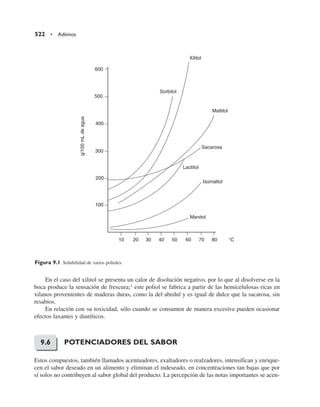
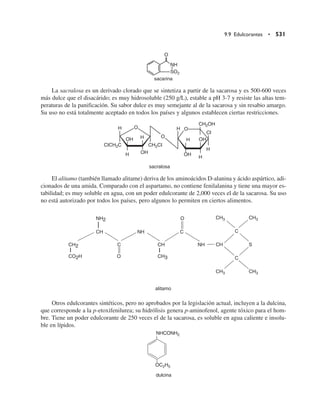
Este documento presenta una revisión de la cuarta edición del libro "Química de los alimentos". Se actualizaron todos los componentes de los alimentos como agua, hidratos de carbono, proteínas, lípidos, vitaminas y minerales. Se incluyeron dos nuevos capítulos sobre tóxicos en alimentos y alimentos transgénicos. El libro es revisado por expertos de reconocidas instituciones y mantiene su estructura general, pero actualizado en cada sección.



















































































































































![Su carácter anfotérico les confiere la capacidad de recibir y de donar electrones y alcanzar un
punto isoeléctrico (pI) cuando presentan el mismo número de cargas positivas y negativas, por lo que
su carga neta es cero. Los aminoácidos pueden tener, por lo tanto, tres estados que dependen del pH:
a pH , pI se encuentran en forma protonada o catiónica; en el pI su carga es cero, y a pH . pI ad-
quieren una carga negativa o aniónica. Es decir, no existe un pH en el cual estos anfolitos estén com-
pletamente ausentes de cargas eléctricas, ya que las presentan aun en el pI. En cualquiera de estos
tres estados pueden ejercer fuerzas electrostáticas. El cuadro 3.1 muestra los valores del punto iso-
eléctrico de los aminoácidos.
La ionización de los aminoácidos es similar a la de cualquier otra molécula, y por lo tanto sigue
la ecuación de Henderson-Hasselbalch:
donde pK, por definición, es el logaritmo negativo de la constante de disociación (K) del grupo ioni-
zable:
Los valores del pK de las distintas funciones ionizables de los aminoácidos se incluyen en el cuadro
3.1. Por ejemplo, el pK del carboxilo de la glicina es de 2.34, ya que a valores de pH menores de 2.34
este grupo se encuentra protonado como COOH, y que a pH mayores se encuentra como COO2. El
pK del amino es de 9.78, ya que a pH , pK toma la forma NH3
+ y a pH . pK, aparece como NH2.
Además de los carboxilos y los aminos, otros grupos también ejercen una influencia en el
comportamiento ácido-base de los aminoácidos: el imidazol de la histidina, el e-amino de la lisina,
el b-carboxilo del ácido aspártico, el sulfhidrilo de la cisteína, el g-carboxilo del ácido glutámico, el
guanidino de la arginina y el hidroxilo fenólico de la tirosina.
El punto isoeléctrico de los compuestos que contienen sólo dos grupos ionizables, un carboxilo
y un amino, se puede calcular a partir de sus respectivos valores de pK:
pI
pK amino pK ácido
2
Aminoácido N base2
H
; K
[base2
] [H
]
[aminoácido]
pH pK log
forma no protonada 1base2
forma protonada 1ácido2
3.2 Aminoácidos • 131
R R R
H H H
NH3
+
NH3
+
(H )
+
(OH )
–
C C C
NH2
COOH COO–
COO–
forma catiónica
(protonada)
forma aniónica
(no protonada)
punto isoeléctrico
(pl) o de doble ion
[Ec. 1]
[Ec. 2]
[Ec. 3]
[Ec. 4]](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-151-320.jpg)










![142 • Proteínas
3.4.1 Reacciones químicas de los grupos funcionales de las proteínas
Reacción con Ninhidrina. Se utiliza a menudo para cuantificar aminoácidos libres, péptidos y pro-
teínas. El aminoácido reacciona con un exceso de ninhidrina [Ec. 5], que le causa una desaminación
oxidativa y la producción de amoniaco, el aldehído correspondiente, que tiene un átomo menos de
carbono que el aminoácido original, CO2 e hidrindantina que proviene de la reducción simultánea de la
ninhidrina. El amoniaco liberado subsecuentemente reacciona con una molécula de hidrindantina,
formando un producto color púrpura conocido como morado de Ruhemanns [Ec. 6], que tiene una
absorbancia máxima a 570 nm. La prolina y la hidroxiprolina dan un producto amarillo que tiene una ab-
sorbancia máxima a 440 nm. Estas reacciones coloridas son la base para las detecciones en cromato-
grafía en papel y de capa fina (TLC), así como para la determinación colorimétrica de aminoácidos que
se utiliza en los analizadores de aminoácidos para lo cual la proteína se hidroliza primero hasta amino-
ácidos libres. Los aminoácidos libres son separados utilizando cromatografía de intercambio iónico/
hidrofóbica. Los eluidos de las columnas reaccionan con ninhidrina y son cuantificados por medición
de absorbancia a 570 y 440 nm. Este método es destructivo, así que el material detectado ya no se
puede utilizar más adelante.
O O
O
O
R
2
O
OH
+ H2N — CH — CO2H
OH
O
O
O
OH
+ RCH + CO2 + NH3
ninhidrina aminoácido hidrindantina
O
O O
OH
N
Reacción con fluorescamina
Los aminoácidos, péptidos y proteínas que contienen aminas primarias producen, con fluorescami-
na, un derivado altamente fluorescente con una máxima emisión a 475 nm cuando la excitación se
hace a 390 nm [Ec. 7]. Este método se puede utilizar para cuantificar aminoácidos, proteínas y pép-
tidos. Es mucho má sensible y no tiene las desventajas de la ninhidrina. El grupo amino que ha reac-
cionado puede ser recuperado con alto rendimiento por hidrólisis ácida y puede ser identificado por
análisis subsecuentes. El exceso de fluorescamina es inestable en presencia de agua, se hidroliza a
productos no fluorescentes y no reactivos, lo que permite que los péptidos detectados con fluoresca-
mina puedan ser analizados directamente. Otra ventaja de este reactivo es su gran sensibilidad, la
cual puede ser utilizada para detectar pequeñas cantidades de proteínas más que con ninhidrina.
[Ec. 5]
[Ec. 6]](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-162-320.jpg)
![Reacción con Orto-ftalaldehído
La reacción de aminoácidos con O-ftalaldehído (1,2-benzeno dicarbonal) en la presencia de 2-mer-
captoetanol produce un derivado fluorescente que tiene una excitación máxima a 380 nm y una emi-
sión de fluorescencia máxima a 450 nm [Ec. 8]. Es tan sensible que es posible cuantificar aminoáci-
dos a niveles de picomoles (10-12 mol).
La ninhidrina, fluorescamina y O-ftalaldehído no son específicos para proteínas porque estos
reaccionan con otros grupos amino.
3.4 Detección y cuantificación de aminoácidos péptidos y proteínas • 143
O
O
O
O
O
fluorescamina
OH
+ R — NH2
R — N
O
O
CH
+ R9SH + RNH2
CH
N — R
SR9
Análisis de los aminoácidos de las proteínas
Este análisis se efectúa por métodos de cromatografía de intercambio iónico con dos resinas, una ca-
tiónica y otra aniónica, con capacidad de separar las moléculas del aminoácido por su afinidad. El
paso previo es la hidrólisis de la proteína a sus aminoácidos constituyentes, en condiciones muy drás-
[Ec. 7]
[Ec. 8]](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-163-320.jpg)


![146 • Proteínas
La reacción de biuret es específica para la medición del enlace peptídico, por lo que sólo se re-
comienda para la cuantificación de proteínas, mas no de hidrolizados, a menos que se conozcan los
tamaños moleculares y se adapte la proteína estándar de la curva. Se utiliza una solución diluida de
sulfato cúprico en tartrato fuertemente alcalino, ésta se adiciona a la solución de proteína, resultan-
do un compuesto de color entre púrpura y violeta que absorbe a 540 nm, obtenido probablemente por
el acomplejamiento del Cu12 con dos enlaces peptídidos adyacentes [Ec. 9].
Se llevan a cabo varias reacciones en condiciones alcalinas y el ión cúprico (Cu12) tiende a oxi-
dar la cadena peptídica reduciéndose a Cu1. Dado que el grupo 2NH del enlace peptídico está in-
volucrado en la reacción, los residuos de Pro no participan y las proteínas con alto contenido de la
misma, producen respuestas bajas. La desventaja de la técnica de biuret es su baja sensibilidad, pues
se requiere de 20-40 mg de proteína para una adecuada detección y existen varios pigmentos que ab-
sorben a 540 nm, longitud de onda de la medición. La presencia de NH4
1 interfiere así mismo con
la reacción. El desarrollo de colores diferente para cada proteína, y los lípidos e hidratos de carbono
interfieren al formar complejos con el ión coordinado.
El ensayo clásico para medir proteínas es el método diseñado por Lowry, Rosebrough, Farr
y Randall llamado Folin-fenol, basado en la reacción de biuret mejorada por la adición del reacti-
vo de Folin-Ciocalteu, cuyos constituyentes activos son los ácidos mezclados fosfomolíbdico-túngs-
tico [Ec. 10].
3H2O • P2O5 • 13WO3 • 5MoO3 • 10H2O
3H2O • P2O5 • 14WO3 • 4MoO3 • 10H2O
Esta mezcla es reducida por la proteína, a través del Cu1 generado y la pérdida de uno o dos
oxígenos de los tungstatos y molibdatos que generan un color azul con absorción máxima de 720-
750 nm. La respuesta del color azul de diferentes proteínas en este ensayo es más variable que en la
reacción de biuret, ya que Tyr, Trp, His y Asn son los residuos que participan en la reacción y Pro,
como en el método de biuret, por su estructura cíclica evita que la cadena peptídica adyacente reac-
cione; este método ha sido modificado muchas veces. En general, la absorbancia se mide a 750 nm
(alta sensibilidad) para proteínas concentradas y se requiere de una curva patrón que es recomenda-
ble hacer con la misma proteína que se cuantifica. La sensibilidad del método va de 0.2 a 300 mg/mL.
Su principal desventaja es que varias sustancias no proteínicas (sacarosa, lípidos, amortiguadores de
pH, monosacáridos y hexosaminas, sulfato de amonio, sulfhidrilos y fosfatos) interfieren produciendo
un color azul o inhibiendo el desarrollo de color por la proteína.
HN
HN
NH
C w O
O w C
NH
R — CH
HC — R
Cu2+
[Ec. 9]
[Ec. 10]](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-166-320.jpg)

![148 • Proteínas
marcado el grupo a-amino. Los dos reactivos más usados son el fluoro-2,4-dinitrobenceno (DNF)
o reactivo de Sanger y el cloruro de dansilo. El a-amino derivado con DNF se colorea de amarillo
vivo. El derivado del cloruro de dansilo, también llamado cloruro de dimetilaminonaftilsulfonilo, se
obtiene en condiciones fuertemente alcalinas, con la ventaja de ser altamente fluorescente.24, 99
La determinación de la composición proporciona una información limitada sobre una proteína,
por lo que es más importante identificar la secuencia de los aminoácidos, para lo que se cuenta con
dos métodos: la forma directa que consiste en llevar a cabo la secuenciación de la cadena polipeptí-
dica en equipos automatizados llamados secuenciadores de proteínas. Por el tamaño de las molécu-
las, puede ser importante hidrolizarlas enzimáticamente con proteasas específicas, para posterior-
mente secuenciar e identificar más fácilmente porciones de 10 a 20 aminoácidos de los péptidos
generados. La combinación de proteasas específicas y los péptidos generados alimentados a bases de
datos que tengan la información de digestiones enzimáticas, permiten ir completando las secuencias
y se ensaya el orden posible de los péptidos al compararlos con bases de datos que tengan la infor-
mación de digestiones enzimáticas provenientes de proteínas cuya secuencia ha sido ya elucidada. El
segundo método permite el análisis con espectrometría de masas de los péptidos generados y sus ma-
sas moleculares respectivas. El sistema más popular es el llamado “MALDI-TOF”.94
NO2
N(CH3)2
N(CH3)2
SO2Cl
NO2
H2N—CH—C—
SO2—NH—CH—C—
O2N O2N
Fluoro-2,4-dinitro-
benceno
Peptidodinitro-
fenílico
péptido
F NH—CH—C—
O
R1
Peptidodansílico
O
R1
O
R1
Cloruro de dansilo
[Ec. 11]](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-168-320.jpg)

















![166 • Proteínas
En el análisis de la termodinámica de la desnaturalización de un sistema proteínico se conside-
ran sólo dos estadios N y D, para la transición entre las moléculas en estado Nativo (N) y desnatura-
lizado (D). Se describe de la siguiente manera:
[N ] m [D] [Ec. 12]
Con una constante de equilibrio:
KD 5 [D] / [N] [Ec. 13]
El valor total de la actividad (Y) detectada en una mezcla de proteínas con una cierta proporción
desnaturalizada, sería:
Y 5 YNfN 1 YDfD [Ec. 14]
Donde f es la fracción de la proteína; YN y YD son las actividades detectables en cada uno de los
estados.
Su balance de masa indica que:
fN 1 fD 5 1 [Ec. 15]
y su constante de equilibrio sería:
K 5 fD / fN [Ec. 16]
de donde se deduce que:
Y 5 (YN 1 YDK) / (11K) [Ec. 17]
con lo que es posible predecir, a una cierta temperatura constante o con una cierta concentración de
desnaturalizante, el comportamiento Y de la proteína, si se conoce la actividad en estado desnatura-
lizado (YD). Para el caso de una enzima, frecuentemente YD 5 0, ya que la enzima no mostraría ac-
tividad biológica si está desnaturalizada, por lo que la expresión se simplificaría a:
Y 5 YN / (11K) [Ec. 18]
Entonces,
K 5 (YN 2 Y) / Y [Ec. 19]
Tm, temperatura de fusión, o de “melting”, es la temperatura en la cual la mitad de las molécu-
las se encuentran desnaturalizadas [D] y la otra mitad en estado nativo [N]. A altas temperaturas
K .. 0 y a bajas temperaturas 0 , K ,1, si Y = YN.
Si Y 5 YNfN + YDfD, entonces de la combinación de [15] y [14],168 resulta la fracción desnatu-
ralizada como:
fD 5 (YN – Y) / (YN – YD) [Ec. 20]](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-186-320.jpg)
![Ahora puede recalcularse la K de equilibrio de la siguiente manera:
K 5 fD/(1 2 fD) 5 fD/fN 5 (YN 2 Y) / (Y 2 YD) [Ec. 21]
Y si se asocia al cambio de energía libre del Gibbs del proceso de desnaturalización mediante la
ecuación:
DG 5 2RT lnK 5 2RT ln [(YN 2 Y)/(Y 2 YD)] [Ec. 22]
es posible graficar 2RT lnK contra la concentración del agente desnaturalizante en la región de la
transición conformacional dando por resultado una línea recta. La KD de desnaturalización y la DGD
en agua pura se calcula del intercepto al origen, es decir, se calcula la energía libre de la desnatura-
lización en presencia de agua o del buffer, en ausencia del desnaturalizante.
3.6.2 Desnaturalización por cambios de temperatura
El caso particular de la desnaturalización térmica se analiza a continuación; partiendo de la ecuación 22:
DG 5 –RT lnK
y la ecuación:
DG 5 DH –TDS [Ec. 23]
entonces:
2lnK 5 [DH/ RT] – [DS/R] [Ec. 24]
y si de la ecuación de van’t Hoff se grafica –lnK vs 1/T, el resultado es una línea recta (figura 3.32)
de cuya pendiente puede calcularse la entalpía DH de la desnaturalización particular que se estudia.
3.6 Desnaturalización • 167
Y o [N]
100%
K = 1
Tm T(C°)
Figura 3.31 Esquema de la desnaturalización típica de una proteína donde Y representa una actividad hipotética
o propiedad de la proteína, o bien, la concentración de la forma nativa de la proteína [N]. K 5 Constante de equilibrio y
Tm 5 temperatura de “melting”.](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-187-320.jpg)




![172 • Proteínas
3.6.6 Desnaturalización con disolventes orgánicos
Se usan solventes orgánicos en el procesamiento de alimentos cuando se desea solubilizar sustratos,
y para desplazar el equilibrio de algunas reacciones enzimáticas al disminuir la concentración de
agua del sistema. Las proteínas disueltas en un sistema acuoso sufren cambios en su estructura tridi-
mensional cuando son trasladadas a un sistema con un solvente distinto. La dirección del cambio, ha-
cia una mayor o una menor solubilización, dependerá del tipo de solvente al que se le traslade. Hay
dos mecanismos principales que explican este tipo de desnaturalización: la unión directa del solven-
te orgánico a la molécula de la proteína y el cambio en la constante dieléctrica del medio.
Al cambiar una proteína a un sistema con disolventes orgánicos se logra un desdoblamiento o
desplegamiento parcial por el rompimiento de las interacciones hidrofóbicas originales, que puede
volver a plegarse mediante interacciones hidrofóbicas y puentes de H que se mantienen, pero en una
estructura distinta de la nativa.
El efecto de la constante dieléctrica sobre la solubilidad de las proteínas disueltas puede expli-
carse mediante la Ley de Coulomb que indica la relación de la fuerza de atracción entre partículas:
F 5 (q1 q2)/Pr2 [Ec. 25]
Donde q es la carga de una partícula, r es la distancia entre las partículas y P es la constante die-
léctrica del medio.
Los sistemas acuosos son los más frecuentes en los alimentos. El agua es un compuesto que po-
see una constante dieléctrica (P) relativamente alta: 78.5, si se compara con otros solventes orgáni-
cos (cuadro 3.9), lo que indica que la atracción entre las partículas con cargas opuestas (que en el ca-
so de las proteínas provienen las positivas de Lys, His y Arg, y las negativas de Asp y Glu) tiene más
dificultades para lograrse en agua, que si se colocan en glicerol, metanol o etanol. Esta dificultad en
la atracción de las partículas cargadas contribuye a la solubilización de la molécula. Por el contrario,
cuando una proteína se coloca en un solvente de baja P la atracción entre las partículas cargadas sobre-
pasa a la energía cinética a la que están sujetas por la temperatura del ambiente, y se insolubilizan,
sobre todo cuando la distribución de las cargas opuestas en la molécula se encuentra en posiciones
opuestas en la molécula, lo que incrementa el momento dipolo de la proteína y favorece la asocia-
ción molecular. Una molécula de proteína en un solvente acuoso logra disolverse mejor si tiene una
distribución de las cargas más uniforme a lo largo y ancho de su superficie, y un menor momento di-
polo.171 Los cambios de P al cambiar de solvente a la proteína, en cuestión, modifican necesariamen-
te la fuerza de atracción entre los residuos de las proteínas y esto explica la precipitación que sufren
las proteínas cuando se añaden solventes miscibles, como metanol o etanol, a las soluciones acuosas
de proteínas porque la atracción entre las partículas cargadas se refuerza.
CUADRO 3.9 Valores de constante dieléctrica e de diversos solventes.
Clase I P Clase II P
Agua 78.5 N-metilformamida 182.4
Glicerol 42.5 Dimetilformamida 36.7
Etilenglicol 37.7 Metanol 32.6
Formamida 109.5 Etanol 24.3
Propanodiol 32 Tolueno 2.4
Etilendiamina 14.2 CCl4 2.2
Ácido fórmico 58.5](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-192-320.jpg)


![que promueven la hidratación de las proteínas y que se unen a ellas de forma débil. Por el contrario,
las que se unen fuertemente a las proteínas desestabilizan porque no logran hidratarlas eficientemen-
te. El mecanismo involucra la capacidad de las sales para promover el ordenamiento de las molécu-
las del agua que no está íntimamente ligada a la matriz de las proteínas sino al agua que se encuentra
en el seno del líquido rodeando a la proteína (agua “bulk”), por la formación de puentes de H a su al-
rededor, lo que promueve la estabilización de las proteínas. En contraste, las sales que desnaturalizan
a las proteínas rompen la estructuración del agua “bulk” convirtiéndola en un mejor solvente para las
moléculas apolares. En otras palabras, el efecto desnaturalizante de las sales caotrópicas está relacio-
nado con la desestabilización de las interacciones hidrofóbicas intramoleculares y la estabiliza-
ción de esas zonas hidrofóbicas ahora expuestas con las moléculas de agua y las sales que rompen
estructura.
El efecto neto en la solubilidad de las proteínas dependerá de la naturaleza de la proteína y de la
sal utilizada, obteniéndose una solubilización o “salting-in” cuando la presencia de los iones ayuda
a la disolución de la molécula, o bien, de “salting-out”, cuando se precipitan otro tipo de proteínas.
Las sales de iones divalentes como Mg Cl2 y (NH4)2SO4 son los más utilizados (figura 3.36).
3.6 Desnaturalización • 175
Log S
Log S = a – b [sal]
M = b
salting
in
salting
out
[sal]
a
Figura 3.36 Efectos de la concentración de sales en la solubilidad de las proteínas](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-195-320.jpg)
![176 • Proteínas
Log S 5 a 2 b [sal] [Ec. 26]
Donde: a 5 ordenada al origen o solubilidad teórica del polímero sin efecto de la sal.
b 5 independiente de T, pH o naturaleza de la proteína, sólo depende de la naturaleza de la
sal utilizada.
El uso de sales para promover la cristalización de proteínas sigue siendo una técnica muy utili-
zada en cristalografía de rayos X, donde se requiere la formación de cristales altamente ordenados y
no la formación de precipitados amorfos, lo que requiere de paciencia y tiempo para lograr la cali-
dad apropiada de cristales.157
3.6.8 Inactivación mecánica
Las proteínas, sobre todo las proteínas multiméricas son fuertemente afectadas por las fuerzas cor-
tantes o por la agitación. Es el caso de los esfuerzos cortantes que se ejercen por ejemplo al bombear
una solución proteínica a través de tuberías, donde al elevar el flujo se desdoblan las estructuras
oligoméricas y el rompimiento de algunos enlaces puede acarrear desnaturalización. El caso de la
agitación y mezclado, que son operaciones unitarias comúnmente aplicadas para la solubilización de
sustratos o para mantener en suspensión algún componente del proceso, no necesariamente implica
esfuerzos cortantes, pero sí la formación de interfases líquido–aire en procesos como el espumado
cuando la proteína se desdobla y se adsorbe, por lo que estabiliza las burbujas de aire formadas. Para
evitar este tipo de desnaturalización es necesario utilizar antiespumantes.
3.6.9 Proteólisis
En términos estrictos, la proteólisis no está considerada como una desnaturalización, ya que se frag-
menta con ella el esqueleto polipeptídico y por lo tanto no se trata de un cambio de conformación,
sino de un rompimiento irreversible de enlaces covalentes de la proteína. Así mismo, durante las ope-
raciones del procesamiento de alimentos es frecuente encontrar consecuencias más severas para la
estructura proteínica, como cuando se involucran la degradación proteolítica por la acción de protea-
sas que se encuentren como impurezas en los tejidos o en las preparaciones enzimáticas. En la natu-
raleza, la desactivación de proteínas llamada “recambio” tiene por objeto la renovación de las molécu-
las con actividad biológica, o bien para controlar la concentración enzimática en algún sistema, lo
cual se logra precisamente a través de una degradación proteolítica. El recambio también evita la pre-
servación de posibles errores de transcripción o traducción en la síntesis de proteínas, de forma que
se puede corregir gracias a la relativamente corta vida de algunas enzimas fisiológicas como la orni-
tina descarboxilasa de rata que tiene una vida media de 11 min, la catalasa que perdura 30 horas o
bien la lactato deshidrogenasa que permanece en el organismo alrededor de cuatro días.29
MODIFICACIONES QUÍMICAS
El procesamiento de los alimentos frecuentemente induce cambios en las proteínas a través de las
operaciones que involucran la aplicación de calor para cocer, evaporar, secar, pasteurizar o esterili-
3.7](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-196-320.jpg)





![182 • Proteínas
P 1 ?OH S P? 1 H2O [Ec. 27]
P? 1 P? S P 2 P [Ec. 28]
Otra forma de entrecruzamientos no naturales es la causada por el intercambio S-S que ocurre
por calentamientos moderados (70-0ºC) causantes también de polimerización de las moléculas pro-
teínicas, pero no daña el valor nutrimental de los aminoácidos indispensables.
CUADRO 3.11 Valores de LAL en alimentos
Alimento LAL (mg/g proteína)
Papas fritas 390
Pretzels 500
Harina maíz sin nixtamalizar 560
Tortillas de maíz 200
Taco shells (tostadas) 170
Fórmula láctea infantil 150-640
Leche evaporada 590-860
Leche UHT 160-370
Leche HTST 260-1030
Leche secada por aspersión 0
Leche descremada evaporada 520
Análogo de queso 1070
Clara de huevo en polvo 160-1820
Caseinato de calcio 370-1000
Caseinato de sodio 430-6,900
Caseína ácida 70-190
Proteína vegetal hidrolizada 40-500
Agente para batido 6,500-50,000
Aislado de proteína de soya 0-370
Extracto de levadura 120
Fuente: Swaisgood y Catignani, 1991.
3.7.5 Reacciones de las proteínas con agentes oxidantes
Los aminoácidos más lábiles son Met. Cys . Trp cuando se aplican agentes oxidantes, frecuente-
mente utilizados para inhibir crecimiento microbiano en los procesos de alimentos: peróxido de hi-
drógeno e hipoclorito de Na. Met se transforma en sulfóxido de Met al añadir un átomo de oxígeno
y en Met-sulfona al añadir dos de ellos. En caso extremo es posible formar ácido homocisteico (con-
tiene un metileno más que el ácido cistéico) por la desmetilación de Met oxidada. En el caso de Cys
y Cistina, se forman también una serie de compuestos que gradualmente adquieren átomos de oxíge-
no: Cys-sulfénico, Cys-sulfínico y Cys-sulfónico o ácido cisteico. A partir de la Cistina se forma Cis-
tina mono o disulfóxido y Cistina disulfona (figura 3.42).](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-202-320.jpg)

![184 • Proteínas
Otra fuente de agentes oxidantes son los lípidos peroxidados provenientes de alimentos en
proceso de enranciamiento. Otro de los aminoácidos que se daña por procesos oxidativos es la Tyr
cuando se aplican peróxido de hidrógeno y peroxidasa produciendo ditirosina, que se encuentra en
proteínas al natural como la keratina, elastina y colágeno.
3.7.6 Reacciones con nitritos
Las N-nitrosaminas, consideradas entre los cancerígenos más importantes, se forman por la reacción
entre nitritos y aminas secundarias, y en un menor grado aminas primarias y terciarias. Los aminoá-
cidos libres son aminas primarias y pueden reaccionar con los nitritos en los procesos de alimentos
que se llevan a cabo a pHs ácidos y con tratamientos térmicos.
En la producción de embutidos es indispensable la adición de nitritos con el fin de evitar el cre-
cimiento de C. botulinum y la consecuente producción de su toxina, así como para la obtención del
color rosado en las carnes curadas y embutidos. Los nitritos producen ácido nitroso que fácilmente
reacciona con los residuos de Trp, Arg, Tyr y Cys principalmente para formar N-nitrosotriptofano,
con Cys para obtener S-nitrosocisteína y con Pro para formar N-nitrosopirrolidina.29 En embutidos,
el mecanismo más favorecido es la reacción del ácido nitroso con los residuos de aminoácidos que
actúan como aminas primarias, excepto Pro que puede considerarse con una amina secundaria.99 Las
glicosilaminas producidas en las reacciones de Maillard (productos de Amadori y de Heyns) reaccio-
nan fácilmente con los nitritos. En un esfuerzo por minimizar la producción de la N-nitrosaminas al
cocinar, asar a la parrilla o al fuego los embutidos, se añaden ácidos ascórbico y eritóbico para con-
trolar la reacción.
3.7.7 Reacciones con sulfitos
En general, la adición de sulfito en los alimentos previene el oscurecimiento de Maillard, así como
el oscurecimiento enzimático, pero logra reducir puentes disulfuro para producir la versión sulfona-
tada de Cys involucrada en el S-S.
P-S-S-P 1 SO322 P-S- SO322 1 P-S [Ec. 29]
Los puentes disulfuro se restauran en las condiciones ácidas del estómago, además la Cys sufo-
natada no pierde su valor nutricional, aunque sí cambian las propiedades funcionales por la pérdida
de estructura con la ruprtura del S-S.29
3.7.8 Reacciones carbonil amino
Estas reacciones, más conocidas como reacciones de oscurecimiento no enzimático o reacciones
de Maillard, se presentan en los alimentos sometidos a altas temperaturas como el horneado, y que
tienen un alto contenido de proteínas y carbohidratos, de forma que involucran al grupo aldehído re-
ductor de los azúcares (aldosas y cetosas), de sustancias como el ácido ascórbico y de los carbonilos
producidos por el enranciamiento de lípidos, y a los grupos amino libres de los aminoácidos, frecuente-
mente el e-amino de Lys. Esto trae consigo una disminución en el valor nutrimental de los alimen-
tos, principalmente al disminuir la biodisponibilidad de Lys, que dependerá de la etapa particular del
oscurecimiento: la base de Schiff que se genera en etapas tempranas de la reacción, se descompone
en Lys y el azúcar en el medio ácido del estómago. A partir de las etapas de la formación de la ceto-](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-204-320.jpg)
![3.7 Modificaciones químicas • 185
samina, es decir de los compuestos de Amadori y el rearreglo de Heyns a aldosamina, Lys pierde su
biodisponibilidad. Posteriormente en la reacción se forman productos de condensación coloridos o
melanoidinas, compuestos insolubles indeseables que se generan durante el almacenamiento de pro-
ductos como la leche en polvo. Sin embargo, hay casos en los que las reacciones coloridas tienen
efectos deseables en alimentos como el pan, las galletas y otros productos en los que también los al-
dehídos resultantes y volátiles forman parte del aroma característico. Estas reacciones deseables ocu-
rren también en los dulces de leche y otros productos de confitería y galletería.
La degradación de Strecker es uno de los mecanismos que forma parte de las reacciones de Mai-
llard. Proviene de la reacción de un aminoácido con un derivado a-dicarbonilo y que termina con la
descomposición del aminoácido en un grupo aldehído, amoniaco y dióxido de carbono.29 Cada alde-
hído da un aroma particular dependiendo del aminoácido que lo origina.
Otra fuente alternativa de grupos aldehídos, en sustitución de los azúcares, proviene del enran-
ciamiento de los lípidos. El aldehído difuncional malonaldehído uno de los más importantes ya que
es capaz de entrecruzar a las proteínas causando insolubilización, pérdida de digestibilidad, y conse-
cuentemente una disminución de la biodisponibilidad.
PROT – NH2 + OHC – CH2 – CHO S PROT – N = CH – CH2 – CH = N – PROT
Siendo un poderoso agente involucrado en el daño de los alimentos durante el enranciamiento
de grasas, es uno de los indicadores más utilizado y se cuantifica utilizando ácido tiobarbitúrico.
De forma indirecta, durante las reacciones de Maillard se oxidan especialmente Met, Tyr, His y
Trp a causa de la presencia de carbonilos insaturados y radicales libres provenientes del proceso de
enranciamiento.
3.7.9 Formación de acrilamida en altas temperaturas
En 2002, investigadores suecos encontraron niveles significativos de acrilamida, compuesto altamen-
te neurotóxico, teratógeno y mutágeno en productos ricos en carbohidratos, como pueden ser papas
a la francesa, papas fritas, botanas, pan, pan tostado, etcétera (cuadro 3.12) como resultado de los
procesos térmicos realizados a altas temperaturas
Los mecanismos propuestos implican a Asn como el aminoácido involucrado a través de reac-
ciones de Maillard para formar un compuesto de Amadori que reacciona fácilmente con los extre-
mos reductores de los almidones presentes hasta formar efectivamente la acrilamida.156 (Figura 3.44).
Hay evidencias de que una disminución en los niveles de Asn permite reducir la formación de
acrilamida y ya existen investigaciones tendientes a producir por ingeniería genética variedades
de papas bajas en ese aminoácido. Asimismo, se ha encontrado que en los procesos tradicionales don-
de se fermenta la masa para el leudado se reducen significativamente los niveles de Asn disponibles
para la posterior reacción química en el horneado, gracias a la fermentación ya que la levadura con-
sume el aminoácido.49 Las prácticas modernas de panificación en las que se alcanzan los niveles de
azúcares fermentables utilizando enzimas, y no por fermentación, han desplazado a los métodos tra-
dicionales y actualmente se generan niveles altos de acrilamida en el pan.
Los niveles medios de exposición38 se encuentran por debajo de 1 mg/kg de peso /día en adul-
tos (con valores que van de 0.28 a 0.71 mg/kg de peso/día) y cerca de 1.5 veces más para el caso de
[Ec. 30]](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-205-320.jpg)




![190 • Proteínas
Propiedades relacionadas con interacciones proteína-proteína. Se trata de las propiedades de pre-
cipitación, gelación, formación de estructuras como pueden ser la formación de masa, de fibras, de
películas, la adhesión y la cohesión.
Propiedades de superficie. Dependen en forma importante de la composición superficial de la pro-
teína, puesto que de acuerdo a la misma dependerá la capacidad de ligar grasas y sabores. La emul-
sificación y el espumado son dos propiedades relacionadas más directamente con los fenómenos de
superficie.
En realidad, estos grupos de propiedades están interrelacionados; por ejemplo, la gelación invo-
lucra no solamente interacciones proteína-proteína sino también proteína-agua, en tanto la viscosi-
dad y la solubilidad dependen de las relaciones entre proteína-agua y proteína-proteína.
3.8.1 Propiedades de hidratación
El agua es un componente esencial de los alimentos y modifica las propiedades fisicoquímicas de las
proteínas, por ejemplo, al ejercer un efecto plastificante sobre las proteínas amorfas o semicristalinas
modifica su temperatura de transición vítrea y de fusión (TD). La transición vítrea se refiere a la con-
versión de un sólido vidrioso amorfo a un estado flexible plastificado, en tanto que la temperatura de
fusión se refiere a la transición de un sólido cristalino a una estructura desordenada.29
La dispersabilidad, la humectabilidad, el hinchamiento, la solubilidad, el “espesamiento” o au-
mento en viscosidad, la capacidad de atrapamiento de agua, la gelación, la coagulación, la emulsifi-
cación y el espumado, dependen todas de las interacciones proteína-agua. Las moléculas de agua se
unen a diferentes grupos en las proteínas, como los grupos cargados mediante interacciones ion-di-
polo. Así mismo, se unen al esqueleto del enlace peptídico, a los grupos amida de Asn y Gln, y al
grupo hidroxilo de los residuos Ser, Thr y Tyr por interacciones dipolo-dipolo. En el caso de unión a los
residuos no polares se induce una interacción dipolo-dipolo, o bien una “hidratación” hidrofóbica.
La capacidad para ligar agua de las proteínas se expresa como los gramos de agua por gramo
de proteína cuando una proteína en polvo es equilibrada con vapor de agua a una humedad relativa del
90 al 95%. Se relaciona con la composición de aminoácidos: si hay una mayor concentración de ami-
noácidos cargados la capacidad de hidratación es mayor y puede ser calculada a partir de su compo-
sición de aminoácidos usando la siguiente ecuación empírica.84
a 5 ƒ C 1 0.2 ƒ N [Ec. 31]
Donde a es gramos de agua/gramo de proteína, y ƒC, ƒp y ƒN son las fracciones de los residuos
cargados, no polares, y polares, respectivamente. No obstante, el modelo es válido para proteínas mo-
noméricas, ya que en el caso de las oligoméricas se presenta una interfase entre subunidad y subuni-
dad, lo que provoca que los resultados experimentales sean mayores que los predichos por la ecuación.
En la mayoría de las proteínas se cuenta con una monocapa de agua con una actividad acuosa
(aW) de 0.05-0.03, en tanto las multicapas de agua se forman en un rango de actividad de agua de 0.3-
0.7. El agua presente en la monocapa se asocia primariamente con los grupos iónicos, no se congela
y no toma parte en las reacciones químicas como solvente: se le denomina agua “ligada” o tipo I. En
una aW 5 0.9, las proteínas ligan arriba de 0.3-0.5 g agua/g de proteína. Los factores ambientales co-
mo pH, fuerza iónica, tipo de sales, temperatura y conformación de la proteína, influyen sobre la ca-
pacidad de ligar agua de las proteínas. Las proteínas presentan la menor capacidad de hidratación en](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-210-320.jpg)








![3.8 Propiedades funcionales de las proteínas • 199
ductos, las proteínas son los principales agentes con actividad superficial que ayudan en la formación
y estabilización de la fase gaseosa dispersa. Generalmente, las espumas estabilizadas por proteínas
se forman por burbujeo, batido, o agitación de una solución proteínica.
Las propiedades espumantes son evaluadas por diferentes principios. La capacidad espumante
de una proteína se refiere a la cantidad de área interfacial que puede ser creada por la proteína, que
se puede expresar en diversas formas, como sobrerrendimiento o poder de espumado.77, 79
[Ec. 32]
El poder espumante, FP, se expresa como:
[Ec. 33]
El poder espumante generalmente aumenta con la concentración de proteína hasta un valor má-
ximo. Esto se ve afectado por el método que se utilice para formar la espuma. Al igualar las concentra-
ciones de diversas proteínas se obtiene su FP y así se pueden comparar sus propiedades espumantes,
que se ilustran en el cuadro 3.15.
CUADRO 3.15 Comparación del poder espumante de proteínas en solución
Poder espumante a una concentración
Tipo de Proteína de proteína al 0.5% (w/v)
Albúmina Sérica Bovina 280 %
Aislado proteínico de Suero 600 %
Albúmina de huevo 240 %
Ovoalbúmina 40 %
Plasma bovino 260 %
b-Lactoglobulina 480 %
Fibrinógeno 360 %
Proteína de Soya (hidrolizada enzimáticamente) 500 %
Gelatina (obtenida por proceso ácido de piel de puerco) 760 %
Cálculos realizados con base en la ecuación [32].127
Estabilidad de la espuma. Se refiere a la capacidad de la proteína para estabilizar la espuma contra
la gravitación y el estrés mecánico y se expresa como el tiempo necesario para que se drene el 50%
de líquido de una espuma o para la reducción del 50% del volumen de la espuma. Estos métodos son
empíricos y no proporcionan información fundamental sobre los factores que afectan la estabilidad
de la espuma. La medición más directa de la estabilidad de una espuma es la reducción del área in-
terfacial de la espuma en función del tiempo. Los métodos están descritos por Yu (1991)176 y Zhu
(1984).179
FP
Volumen del gas incorporado
Volumen del líquido
3 100
Sobrerrendimiento
Volumen de espuma – Volumen del líquido inicial
Volumen del líquido inicial
3 100](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-219-320.jpg)


![202 • Proteínas
ractuar con las proteínas vía puentes de hidrógeno e interacciones electroestáticas. Después de la
unión a las regiones hidrofóbicas de la superficie, los aldehídos y las cetonas pueden ser capaces de
difundirse al interior hidrofóbico de la molécula de proteína. Los aldehídos pueden unirse covalente-
mente al grupo amino de la lisina, y esta interacción no es reversible, aunque la mayor parte de las
uniones que pueden contribuir al aroma y sabor de un producto proteínico son no covalentes.
La difusión de los compuestos de sabor en el interior de la proteína puede romper las interaccio-
nes hidrofóbicas entre los segmentos de proteínas, y por lo tanto desestabilizar la estructura de la pro-
teína. El sabor que se une a los grupos reactivos, como los aldehídos, puede unirse covalentemente
al grupo e-amino de los residuos de lisina, lo que modifica la carga neta de la proteína y, por lo tan-
to, causa el desplegamiento de la proteína. Este fenómeno resulta en una exposición de nuevos sitios
hidrofóbicos para la unión de ligandos. En el caso de proteínas oligoméricas, como las de soya, los
cambios conformacionales pueden involucrar tanto disociación como desplegamiento de subunida-
des. Las proteínas desnaturalizadas generalmente presentan un gran número de sitios de unión con
constantes de asociación débiles.
Factores que influyen en la unión del sabor
Los sabores volátiles interactúan con las proteínas hidratadas principalmente vía interacciones hidro-
fóbicas y por lo tanto, cualquier factor que las afecte, o que afecte la hidrofobicidad de la superficie,
a su vez influirá en la unión del sabor.158 La temperatura tiene poco efecto en el proceso de unión al
sabor, a menos que ocurra un desplegamiento térmico de la proteína. Esto sucede porque el proceso
de asociación está impulsado por entropía y no por entalpía. Las proteínas desnaturalizadas térmica-
mente presentan una mayor capacidad para unir sabores; sin embargo, la constante de unión es gene-
ralmente más baja comparada con la proteína en estado nativo. El efecto de las sales sobre la unión
del sabor se relaciona con las propiedades de “salting-in” y “salting-out”, ya que las sales que pro-
mueven la solubilización desestabilizan las interacciones hidrofóbicas y bajan la unión al sabor, y
ocurre lo contrario con las sales que producen “salting-out”. El pH afecta la unión del sabor porque
tiene efectos sobre la conformación, favoreciendose la unión al sabor a valores de pH alcalinos más
que a ácidos, ya que la proteína tiende a desnaturalizarse más fácilmente en los primeros.
3.8.4 Viscosidad
La aceptación de alimentos líquidos y semisólidos por parte de los consumidores de productos tipo
salsas, sopas, bebidas, etcétera, depende de la viscosidad y consistencia del producto. La viscosidad,
que se define como la resistencia de una solución a fluir bajo una fuerza aplicada o un esfuerzo de
cizalla. Para el caso de una solución ideal,85 el esfuerzo de cizalla es directamente proporcional a la
velocidad de corte. Es decir, que la fuerza por unidad de área, F/A, es directamente proporcional al
gradiente de velocidad entre las capas del líquido, dv/dr, expresada como:
[Ec. 34]
La constante de proporcionalidad h se conoce como el coeficiente de viscosidad. Los fluidos que
obedecen esta expresión son llamados fluidos Newtonianos. La conducta del flujo de las soluciones
se afecta principalmente por el tipo de soluto. El alto peso molecular de los polímeros solubles incre-
F
A
h
dv
dr](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-222-320.jpg)





































































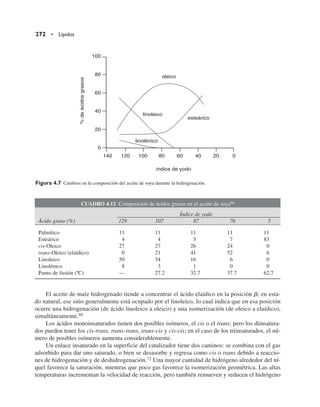













![En los hidroperóxidos del oleico se rompe la unión O–O y se sintetiza el radical alcoxi corres-
pondiente, para después escindir el enlace C–C en dos posiciones, a la derecha y a la izquierda, lo
que produce una gama enorme de compuestos de bajo peso molecular (cuadro 4.15).
286 • Lípidos
—CH CH—CH2—CH CH—
—CH CH—CH—CH CH— (I)
[R•]
•
•
[RH]
—CH—CH CH—CH CH— (II) —CH CH—CH CH—CH— (III)
O2
O
O•
[ROO•] [ROO•]
RH
R•
O2
•
—CH—CH CH—CH CH— (IV) —CH CH—CH CH—CH— (V)
O
O•
RH
R•
13 12 11 10 9
13 12 11 10 9
—CH—CH CH—CH CH— (VI) —CH CH—CH CH—CH— (VII)
13 12 11 10 9
O
OH
O
OH
Figura 4.13 Mecanismo de oxidación del ácido linoleico.](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-306-320.jpg)


























![de alimentos, son importantes para la biotecnología moderna. Éste es el caso de la ADN polimerasa
obtenida de Thermus aquaticus, esencial para llevar a cabo la reacción en cadena de la polimerasa
(PCR); o de lipasas obtenidas de Thermotoga spp. Las enzimas termófilas extremas tienen un poten-
cial de aplicación en el campo alimentario si consideramos que se necesitan enzimas que resistan
condiciones drásticas de operación, por ejemplo, en el procesamiento de almidón o que operen a tem-
peraturas que limiten los riesgos de contaminación microbiana. Es importante recordar que los pro-
cesos químicos se desarrollan generalmente a altas temperaturas.
5.6.3 Efecto de la concentración de sustrato
Una enzima funciona de manera más eficiente cuando la concentración de sustrato está en exceso en
relación con la concentración de enzima. Esto se debe a que las colisiones “exitosas” con el reactivo
son más frecuentes, asegurando así que la mayor cantidad de enzima se encuentre activa. En estas
condiciones, el producto se obtiene a la máxima velocidad posible para la cantidad de enzima pre-
sente. En caso de que la concentración de sustrato sea menor, la velocidad de reacción disminuye ge-
neralmente de acuerdo con un comportamiento como el que se muestra en la figura 5.7. Este aspecto
es muy importante en la caracterización cinética de una enzima, como se verá más adelante. Por otra
parte, la acción de una enzima a nivel industrial debe ser óptima tanto en términos de costo como de
eficiencia catalítica, por lo que la reacción se debe llevar a cabo en la medida de lo posible a la má-
xima velocidad.
314 • Enzimas
Concentración de sustrato
[S]0
Km
Velocidad
inicial
vi
vi =
Vmáx
2
[S]0 Km
vi = Vmáx
vi =
Vmáx [S]0
Km + [S]0
[S]0 Km
vi =
Vmáx [S]0
Km
Zona de orden cero
Zona de
orden uno
Figura 5.7 Efecto de la concentración del sustrato en la velocidad de reacción de las enzimas.](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-334-320.jpg)


![Según lo indica el sentido de las flechas, tanto la formación del complejo enzima-sustrato como
su consumo para liberar al producto, pueden ser procesos reversibles. Los coeficientes k1, k1, k2 y
k2 representan las constantes de velocidad para cada reacción. La formación del complejo ES es
generalmente rápida, mientras que su descomposición en producto y enzima libre es un paso lento,
(k2 k1), mientras que k2 es generalmente mucho mayor que k2. Sin embargo, bajo ciertas condicio-
nes, una reacción catalizada enzimáticamente puede proceder en cualquier dirección. Por ejemplo, y
como se ha señalado ya, algunas reacciones enzimáticas que proceden en dirección hacia la hidrólisis en
un medio acuoso, pueden proceder en sentido inverso en un medio con baja disponibilidad de agua.
Durante los primeros instantes de la reacción, la velocidad de formación de producto (∆P/∆t) se
define como velocidad inicial, vi, y es, de acuerdo con la ecuación 3, proporcional a la concentración
del complejo ES. La concentración de enzima total (ET), en cualquier momento es igual a [E] [ES]
(enzima libre enzima en forma de complejo (Ec. 4)). Cuando [S][E] la enzima está totalmente
saturada, esto es, se encuentra formando el complejo con el sustrato (Ec. 5), por lo que se encuentra
a su máxima velocidad (Vmáx), como se expresa en la ecuación 6.
vi k2 [ES] (Ec. 3)
[E]T [E] [ES] (Ec. 4)
Si [S][E], entonces
[E]T ≈ [ES] (Ec. 5)
vi ≈ k2 [E]T Vmáx (Ec. 6)
Es importante recordar que existe una relación entre la concentración de los reactivos con la ve-
locidad de reacción, lo que define el orden de una reacción química. Se ha mencionado que en el ca-
so de una reacción enzimática la concentración de sustrato se debe mantener muy alta con relación a
la concentración de enzima, de tal forma que se trate de una reacción de orden cero (figura 5.7).
Así, la velocidad de reacción, a pH y temperatura constantes, será independiente de la concentración
de sustrato y dependerá solamente de la concentración de la enzima, como lo muestra la figura 5.8 y
la ecuación 6.
5.7 Cinética de las reacciones enzimáticas • 317
concentración de enzima
velocidad
Figura 5.8 Velocidad de reacción enzimática con respecto a la concentración de enzima. La línea punteada es la relación
teórica (ecuación 6), y la línea sólida la relación observada.](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-337-320.jpg)
![Por otra parte, en la figura 5.7 se observa que en concentraciones bajas de sustrato, la velocidad
vi en los inicios de la reacción es proporcional a la concentración de sustrato y, por lo tanto, se esta-
blece un sistema de primer orden; a medida que se incrementa [S], la velocidad se vuelve de orden
fraccionario y posteriormente de orden cero; en este último caso vi es independiente de la concentra-
ción del sustrato, la enzima alcanza su saturación y se obtiene la máxima velocidad, como ya se
mencionó anteriormente.
A la relación entre k1, k1 y k2 se le conoce con el nombre de constante de Michaelis-Menten o Km.
(Ec. 7)
La Km, junto con k2, también conocida como constante catalítica (kcat) o número de recambio,
son específicas para cada enzima y la relación kcat/Km determina la eficiencia catalítica de la enzima
para un determinado sustrato (cuadro 5.7).
La ecuación de Michaelis-Menten (Ec. 8) representa una hipérbola (figura 5.7) y describe la va-
riación del orden de reacción en función de la concentración del sustrato: en condiciones de satura-
ción, la ecuación 8 se transforma en vi Vmáx que representa una ecuación de velocidad de orden ce-
ro. Por otra parte, cuando [S]0Km, entonces la ecuacion de Michaelis se puede simplificar a vi
Vmáx ([S]0/Km), que puede simplificarse como v k9 [S], donde k equivale a una constante de pri-
mer orden, como se señala en la misma figura 5.7. En general:
(Ec. 8)
Aparentemente en la igualdad de Michaelis-Menten no existe ningún término que incluya la con-
centración de la enzima [E]T; sin embargo, ésta se encuentra implícita en Vmáx ya que es igual a
k2[E]T, como se precisó en la ecuación 6.
CUADRO 5.7 Ejemplo de valores de las constantes catalíticas para varias enzimas,
algunas con más de un sustrato
Enzima Sustrato kcat (s–1) Km (M–1) kcat/Km (M.s–1)
Acetilcolina esterasa Acetilcolina 1.4 104 9 105 1.6 108
Anhidrasa carbónica CO2 1 106 1.2 102 8.3 107
HCO3
4 105 2.6 102 1.5 107
Catalasa H2O2 4 107 1.1 4 107
Crotonasa Crotonil-CoA 5.7 103 2 105 2.8 108
Fumarasa Fumarato 8 102 5 106 1.6 108
Malato 9 102 2.5 105 3.6 107
Triosafosfato isomerasa Gliceraldehído 3-fosfato 4.3 103 4.7 104 2.4 108
b-lactamasa Bencil penicilina 2 103 2 105 1 108
Hexocinasa Glucosa 1.5 104
Fructosa 1.5 103
Quimotripsina N-benzoil tirosinamida 2.5 103
N-formil tirosinamida 1.2 102
N-acetil tirosinamida 3.2 102
N-glicil tirosinamida 1.2 101
v1 Vmáxc
[S]
Km [S]
d
Km
k
1 k2
k1
318 • Enzimas](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-338-320.jpg)
![De la ecuación 8 se puede deducir una relación importante, ya que cuando vi 1/2 Vmáx, se ob-
tiene que Km [S]; es decir, la relación de constantes de velocidad definida en la ecuación 7, es igual
a la concentración del sustrato a la cual la velocidad observada en la reacción es la mitad de la velo-
cidad máxima. El valor de Km no es absoluto, depende del pH, la temperatura y del tipo de sustrato,
más no de su concentración.
La Km es un índice de la afinidad que tiene una enzima por un determinado sustrato: los valo-
res bajos indican que la enzima requiere de bajas concentraciones de sustrato para alcanzar la satu-
ración; por el contrario, los valores altos representan enzimas con poca afinidad hacia el sustrato, ya
que es necesaria una elevada concentración de éste para que se saturen. Por ejemplo, en el cuadro 5.7
se observa que la quimotripsina hidroliza varios sustratos con diferentes velocidades, dependiendo de
la afinidad que tenga hacia cada uno de ellos; se observa que la N-benzoiltirosinamida es el mejor
sustrato para esta enzima y que el tamaño del grupo unido a la tirosina determina la afinidad de la
enzima hacia cada sustrato, lo que se refleja en el valor de Km.
CUANTIFICACIÓN DE ACTIVIDAD ENZIMÁTICA
La potencia o actividad de una enzima no puede medirse en términos de su concentración, ya que
puede estar presente pero en forma desnaturalizada y sin funcionalidad; por esta razón se emplea la
Unidad de Actividad Enzimática, definida como la cantidad de enzima que se requiere para transformar
en producto una mmol de sustrato por minuto, en las condiciones óptimas de pH y temperatura; la
concentración de sustrato deberá ser aquella en que la enzima se encuentre actuando con su veloci-
dad máxima, es decir, en condiciones de saturación ([S]0Km). Para la determinación, se debe me-
dir la velocidad inicial de consumo de sustrato (∆S/∆t) o la de aparición de producto (∆P/∆t) bajo
condiciones definidas de pH, fuerza iónica y temperatura: ∆S y ∆P equivalen a la cantidad de sustra-
to y de producto que se transformaron en el intervalo inicial de tiempo ∆t.
Cuando se quiere saber la proporción de enzima con respecto a todas las proteínas que pueden
estar presentes en una preparación se utiliza la actividad específica, definida como las unidades de
actividad de la enzima en relación con la cantidad total de proteína en miligramos; cuanto más pura
sea, mayor será la relación de actividad por miligramo de proteína. Esta forma de expresar la activi-
dad enzimática es de gran utilidad para seguir el grado de purificación de una enzima durante los dis-
tintos pasos de su obtención.
Otro término que se emplea con frecuencia es el número de recambio, que equivale al número
de moléculas de sustrato transformadas en producto, por minuto, por molécula de enzima. En gene-
ral, en la mayoría de las transformaciones que sufren los alimentos se tienen valores del número de
recambio que van desde varios miles hasta millones; por ejemplo, los valores de esta unidad para la
invertasa, la polifenol oxidasa, la lipoxigenasa, la lactasa y la b-amilasa, son de 42,000, 120,000,
180,000, 750,000 y 1,200,000, respectivamente. El número de recambio equivale a la actividad espe-
cífica: moles de sustrato transformadas por minuto por cada mol de enzima. Por ello, con frecuencia
sólo se reporta como min1 y equivale a la kcat.
Muchos de los sustratos en alimentos son polímeros de composición química y peso molecular
diverso (por ejemplo, almidón, pectinas, complejos proteínicos) por lo que es complicado definir su
actividad en los términos anteriormente expuestos. Se han desarrollado innumerables métodos para
5.8 Cuantificación de la actividad enzimática • 319
5.8](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-339-320.jpg)
![cuantificar la actividad en esos sistemas, siendo relevantes los casos de las actividades amilolítica y
proteolítica; sin embargo, los protocolos son muy arbitrarios, lo que dificulta la comparación entre
los valores obtenidos por diversos métodos. Por ejemplo, para determinar actividad amilolítica se
cuenta con las siguientes metodologías y unidades de actividad correspondientes:40
• Poder diastásico (DP°). Cantidad de enzima presente en 0.1mL de una preparación enzimáti-
ca al 5% que produce la cantidad de azúcar que reduce 5mL de solución de Fehling, en 100mL
de una solución al 1% de almidón a 20°C por 1 hora.
• Unidad dextrinógena (DU). Cantidad de enzima que cataliza la conversión de 1mg de almidón
soluble al 1.2% en amortiguador de acetatos pH 5.7, en 10 minutos a 30°C.
• Unidad glucoamilasa (GAU). Cantidad de enzima que libera 1g de glucosa a partir de almi-
dón soluble en 1 hora.
• Unidad SKB. Cantidad de amilasa que hidroliza 1g de dextrina b-límite hasta el punto de reac-
ción negativa con yodo en 1 hora bajo condiciones definidas.
• Unidades “licuefón”. Se determina el tiempo requerido para disminuir un 50% la viscosidad
relativa de una cantidad definida de almidón.
• Unidades NF. Tiempo requerido para que 1g de preparación enzimática hidrolice 100g de al-
midón de una solución al 3.75% a 40°C, hasta el punto de reacción negativa con yodo.
Desde el punto de vista de aplicación industrial de enzimas, es probable que se tenga que elegir
de entre un grupo de enzimas de diversas características para una aplicación dada. De acuerdo a lo
que se ha revisado anteriormente la decisión dependerá de varios factores, como los rangos de pH y
temperatura óptimos de funcionamiento de cada una. Otro factor importante es la estabilidad al pH
y a la temperatura de operación, que no necesariamente coincidirá con los valores óptimos de fun-
cionamiento. Adicionalmente, el valor de la Km permitirá elegir a la enzima que tuviera una mayor
afinidad por el sustrato de interés. Para calcular la cantidad de enzima necesaria para llevar a cabo la
reacción en un tiempo estipulado o el tiempo en que se llevaría a cabo la reacción con una cantidad
determinada de enzima, es necesario emplear la ecuación integrada de Michaelis-Menten que a con-
tinuación se presenta:
k2[E]T t [S]o X Km n (1 X) (Ec. 9)
donde:
(Ec. 10)
So es la concentración inicial de sustrato y S la concentración al tiempo t.
USO INDUSTRIAL DE LAS ENZIMAS
De las miles de enzimas conocidas, sólo algunas decenas se producen a escala industrial, para em-
plearse en la manufactura tanto de alimentos como de materias primas. Cada día aumenta el número
de reacciones industriales que se efectúan por rutas enzimáticas, principalmente debido a la posibilidad
que existe hoy en día de expresar cualquier gene en microorganismos modificados genéticamente.
X
[S]O [S]
[S]O
320 • Enzimas
5.9](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-340-320.jpg)




























![5.10 Revisión de enzimas de importancia en alimentos • 351
Ciclodextrin
Amilosacarasa
glucosil
Levan
Dextran
transferasa
sacarasa
sacarasa
una afinidad muy alta la cadena de amilosa del almidón. Además puede catalizar otras reacciones co-
mo la reacción inversa a la ciclización y la de desproporcionación, que se ilustran en la figura 5.21.48
Las ciclodextrinas que se aplican en alimentos provienen de la reacción con almidón y pueden
tener 6, 7 u 8 residuos de glucosa, a, b, o g-ciclodextrinas, respectivamente. En éstas, los hidroxilos
están localizados en la parte externa del anillo, mientras que los grupos no polares se encuentran en
el centro lo que resulta en una molécula que es soluble en agua, pero que provee un microambiente
hidrofóbico en su centro. Las ciclodextrinas se han utilizado en alimentos como agentes estabilizan-
tes de sustancias lábiles a la oxidación o en la liberación controlada de aromas, así como en emulsio-
nes como mayonesa o margarina. Recientemente se ha aplicado la b-ciclodextrina para remover el
colesterol de la leche y de otros productos.48
5.10.6 Isomerasas
Glucosa isomerasa (EC 5.3.1.5). Es una de las enzimas industriales más importantes en el área
de procesamiento de almidón, cuyo uso data de los años 60s. El sustrato natural de esta enzima es la
D-xilosa, que se isomeriza a D-xilulosa, por lo que su nombre correcto es xilosa isomerasa; en la in-
dustria alimentaria se utiliza para la isomerización de D-glucosa a D-fructosa, reacción muy impor-
CUADRO 5.15 Enzimas que catalizan reacciones de transglicosilación de interés en alimentos48
Número Organismo Molécula Molécula
Nombre de EC productor donadora aceptora Producto Aplicación
2.4.1.5 Leuconostoc Sacarosa (Glucosa)n Dextrana Agente
mesenteroides [-(1-6)] viscosante
NRRL B-512F
Leuconostoc Sacarosa Maltosa Gluco- Agentes
mesenteroides oligosacáridos prebióticos
NRRL B-1299 [-(1-2)]
2.4.1.10 Bacillus Sacarosa (Fructosa)n Levana Agente
polymyxa, [-(2-6)] espesante
Zimomonas Sacarosa Oligosa Fructo- Agentes
mobilis cáridos oligosacáridos prebióticos
[-(2-6)]
2.4.1.19 Bacillus Amilosa Amilosa Ciclodextrinas Vehículo de
macerans, B. (, , o
) moléculas no
megaterium, polares
Klebsiella sp.,
Thermoanaero-
bacter
2.4.1.4 Neisseria Sacarosa Almidón Almidón Almidones
polysaccharea modificado resistentes
no digeribles](https://image.slidesharecdn.com/quimicadelosalimentos-220701160554-7444d8a7/85/Quimica-de-los-alimentos-pdf-371-320.jpg)