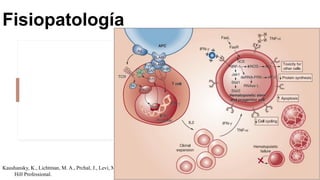
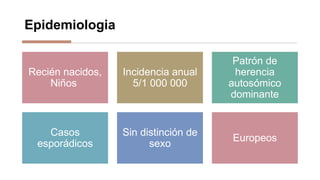
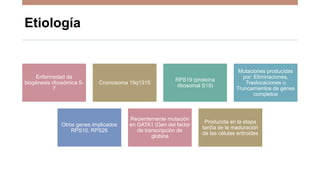
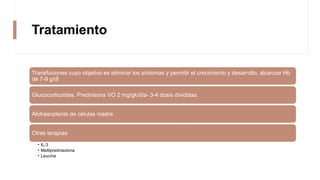
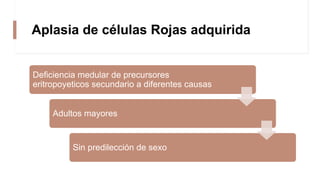
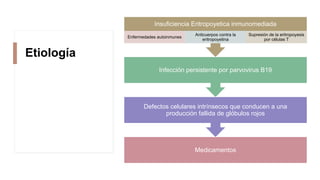
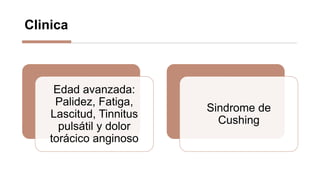
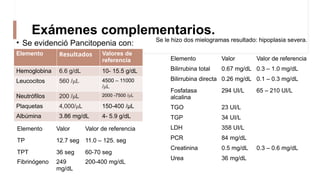
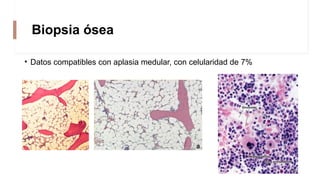
La anemia aplásica es una enfermedad caracterizada por pancitopenia y hipocelularidad medular, con una incidencia en México de 4.2 por millón/año en pediátricos y 3.8 en adultos, predominando en varones. Los síntomas incluyen debilidad, disnea y hemorragias, y el diagnóstico se realiza mediante frotis de sangre y biopsia de médula ósea. El tratamiento abarca desde trasplante de células madre hasta inmunosupresión, con un pronóstico malo para los casos graves.