Descargado 114 veces






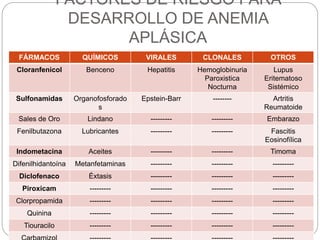
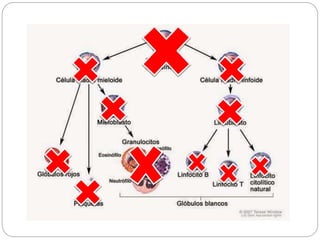

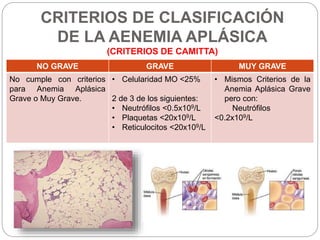

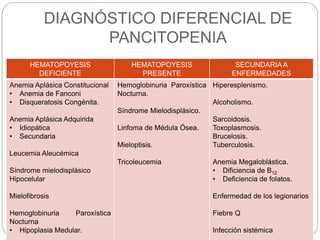








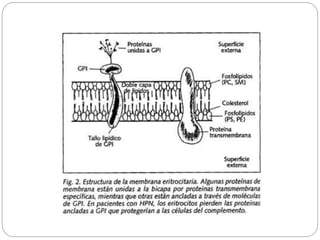








El documento aborda la anemia aplásica y la hemoglobinuria paroxística nocturna, definiéndolas como enfermedades hematológicas que implican alteraciones en las células hematopoyéticas. Se discute su fisiopatología, clasificación, diagnóstico y opciones de tratamiento, incluyendo la terapia inmunosupresora y el trasplante de médula ósea. Además, se exploran factores de riesgo, presentación clínica y pronóstico asociado a estas condiciones.













































































