Descargado 55 veces

















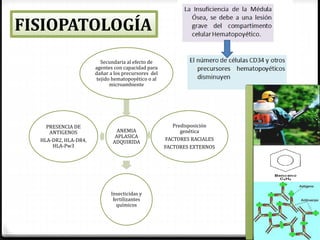





















1. La aplasia medular adquirida es una enfermedad que causa una disminución de las células sanguíneas debido al daño del tejido hematopoyético en la médula ósea, lo que lleva a pancitopenia. 2. Puede ser causada por fármacos, radiaciones, virus u otras enfermedades y suele tratarse con transfusiones de sangre, antibióticos y trasplante de médula ósea. 3. La inmunosupresión con anticuerpos antitimocíticos y cic























































































