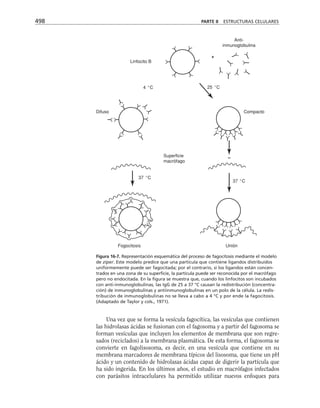
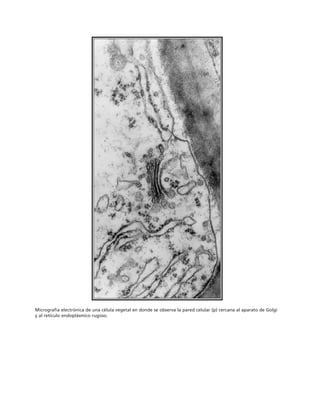
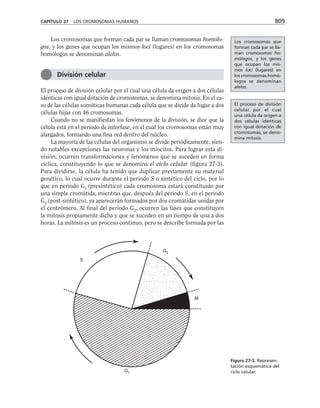
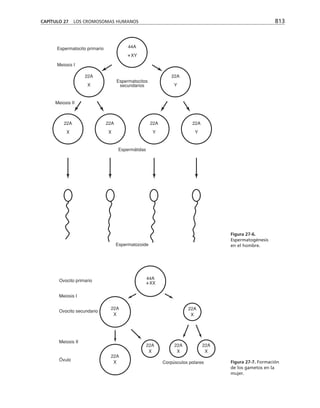
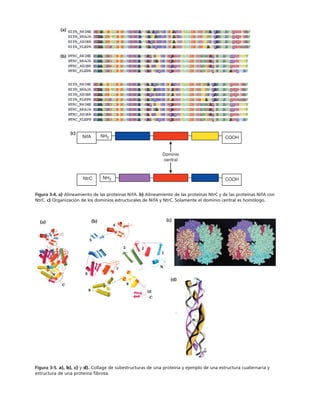
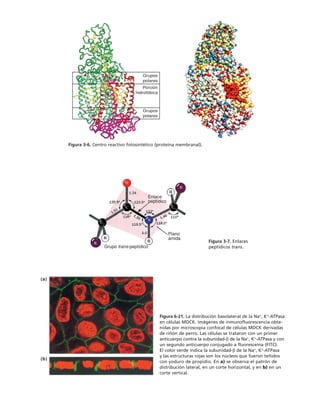
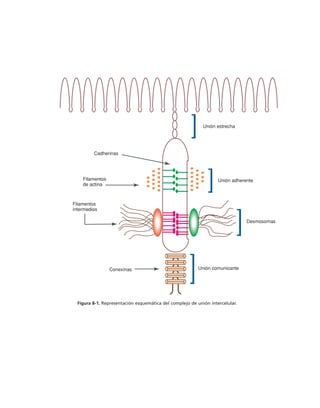
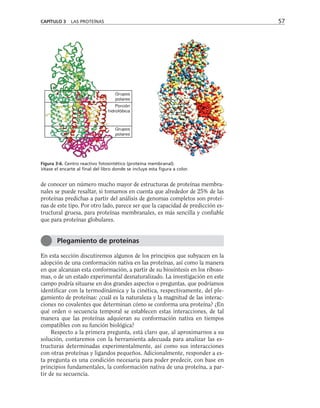
Este documento presenta un libro de texto sobre biología celular y molecular. El libro fue editado por Leticia Gaona Figueroa y coordinado por Luis Felipe Jiménez García y Horacio Merchant Larios de la Universidad Nacional Autónoma de México. Fue publicado por Pearson Educación de México en 2003. El libro contiene seis capítulos que cubren temas como la estructura y función del ADN, ARN y proteínas, así como la membrana celular y los mecanismos moleculares de diferenciación celular.






















































































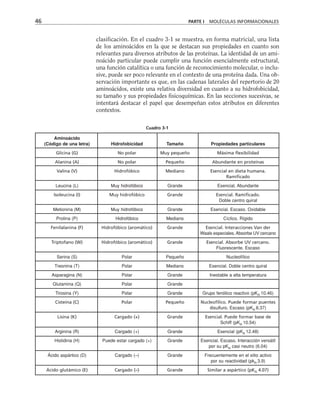
















































































































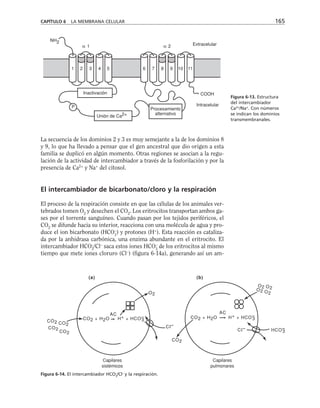

















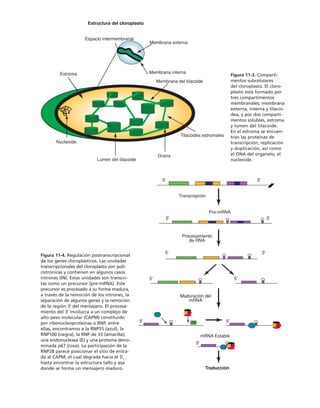
![El flujo de iones a través de los canales constituye una corriente eléc-
trica cuya magnitud y cinética se pueden estudiar utilizando aparatos electró-
nicos, primeramente para amplificar la señal y después para procesarla y re-
gistrarla. Ésta es la razón de que el estudio de canales iónicos requiera que
recordemos aquí algunos conceptos y técnicas electrofisiológicas; el más
fundamental de ellos es la ley de Ohm:
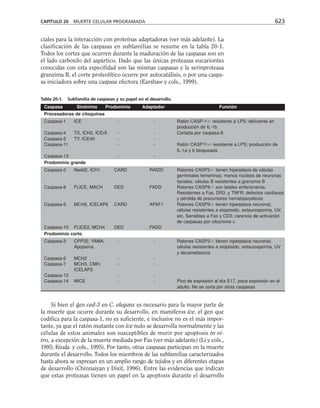
I = g E
Donde I es la corriente en amperios, g es la conductancia en siemens y E es
el potencial eléctrico transmembranal en volts.
Así, en su forma más simple, los canales iónicos se pueden modelar
considerándolos en un circuito eléctrico, como una resistencia, o su inver-
sa que es la conductancia. Cada canal inserto en la membrana tiene su pro-
pia conductancia unitaria ( gi
). Si todos los canales son de un mismo tipo,
la conductancia total de membrana ( gm
) de una célula es la suma de las
conductancias unitarias de los (n) canales abiertos.
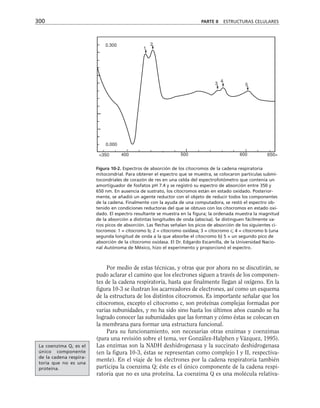
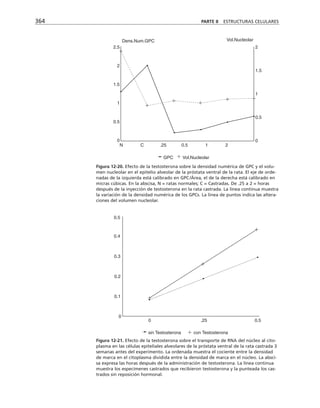
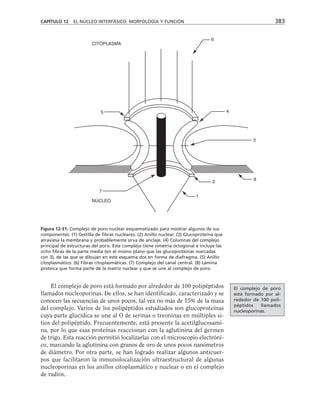
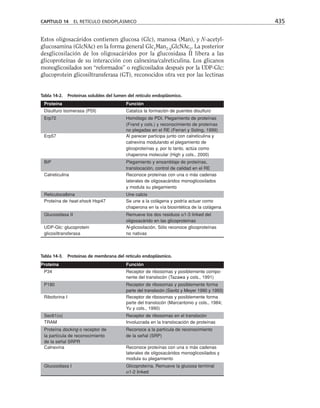
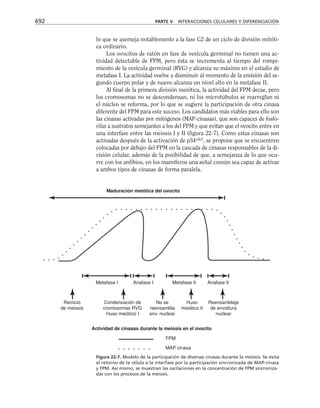
Además, es necesario considerar la membrana celular como un capaci-
tor que separa el medio citoplasmático del exterior celular. Este capacitor
está arreglado en paralelo con la resistencia, de manera que el modelo eléc-
trico de membrana es el siguiente:
CAPÍTULO 6 LA MEMBRANA CELULAR 183
Eion
Rm
Cm
El gradiente electroquímico que impulsa los iones a través de los cana-
les depende de la diferencia de concentración en ambas caras de la membra-
na y de gradiente eléctrico, o la diferencia de potencial entre el interior y el
exterior celular. Cuando el gradiente químico y el eléctrico se encuentran
en equilibrio, los iones no fluyen a través de los canales, aunque éstos se en-
cuentren abiertos. Éste es el potencial de reposo (Eion
), que se puede calcu-
lar con la ecuación de Nernst como sigue:
Eion
= ln
El potencial de reposo es distinto para cada especie iónica, el sentido y
la magnitud del flujo a través de los canales, depende de la diferencia entre
el potencial de membrana y el potencial de reposo para un cierto ion, como
lo describe el siguiente modelo:
Iion
= gion
(Em
– Eion
)
La corriente total de membrana, que se puede medir en un momento
dado, es igual a la corriente capacitiva, más la corriente iónica.
[ion]ext
–
–
–
–
–
–
–
–
–
–
–
–
[ion]ion
RT
–
–
–
–
zF](https://image.slidesharecdn.com/biologia-celular-y-molecular-220613202029-80f4c21c/85/biologia-celular-y-molecular-pdf-224-320.jpg)


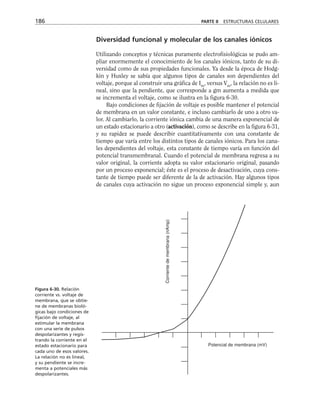
























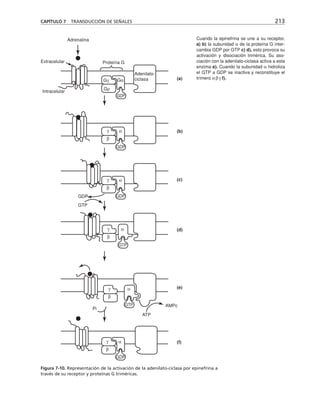





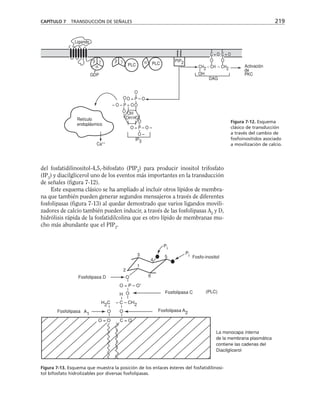























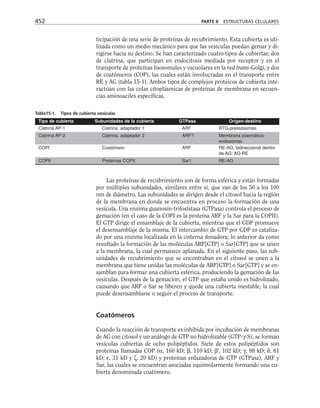
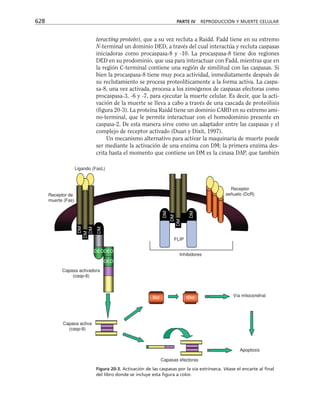
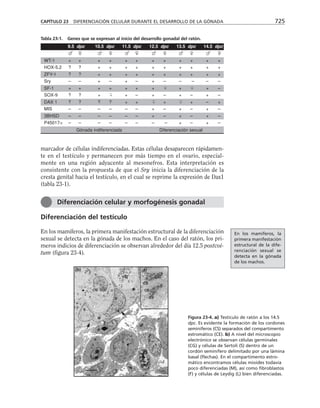
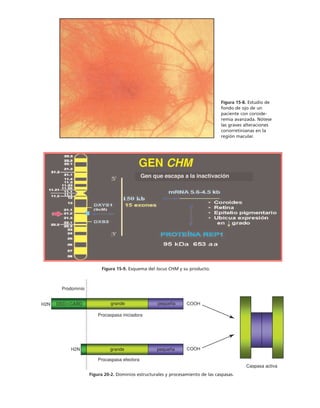
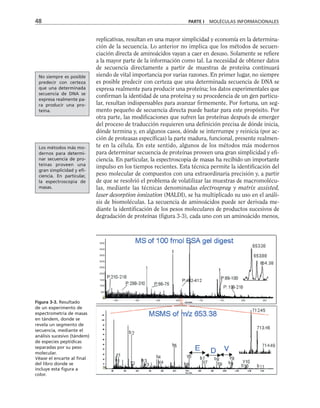
![Participación de la proteína G, la fosfolipasa C
y la proteína-cinasa C en el ensamble y sellado
de las UEs
Durante el proceso de ensamble de las UEs el calcio actúa desde el medio
extracelular a través de las moléculas de adhesión dependientes de calcio
(cadherinas) que describiremos más adelante. Esta señal de contacto inter-
celular se transduce y desencadena la activación secuencial de una proteína
G inhibitoria sensible a la toxina pertussis, la fosfolipasa C y las proteínas
cinasas C (PKC) sensible a diacilglicerol (clásicas y nuevas) (figura 8-10).
Paralelamente se activa la calmodulina, se rearregla el citoesqueleto de ac-
tina y se insertan en la membrana los componentes de la UE almacenados
en los compartimentos vesiculares (Balda y cols., 1991 y 1993).
CAPÍTULO 8 LOS CONTACTOS INTERCELULARES 251
RET
(Ω/cm
2
)
1600
1400
1200
1000
800
600
400
200
0
– + – + – + – + + +
Prot G FLC PKC CaM [AMPc]
1.8
mM
PTX
HLT
H7
diC8
TFPZ
dB–AMPc
Forsk
IBMX
5
µM
Neomicina
Al
F
3
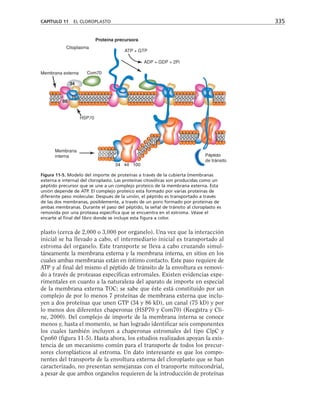
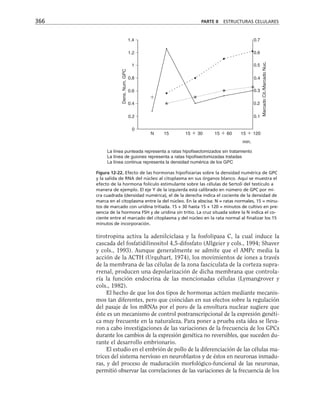
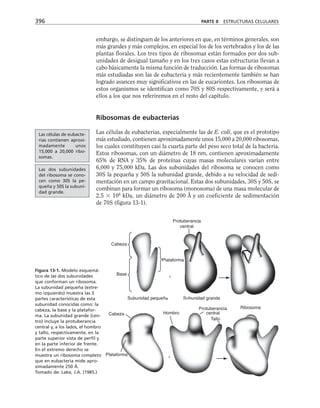
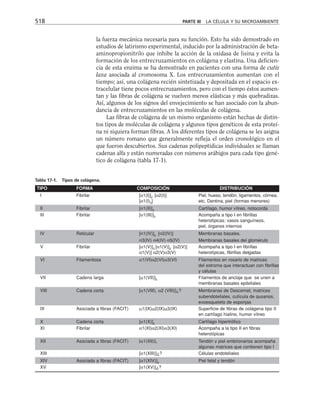
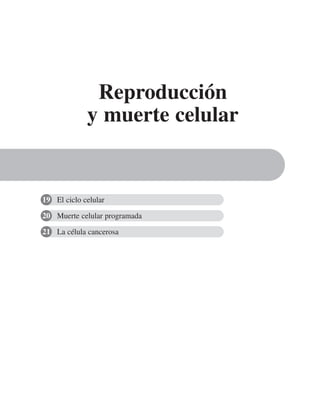
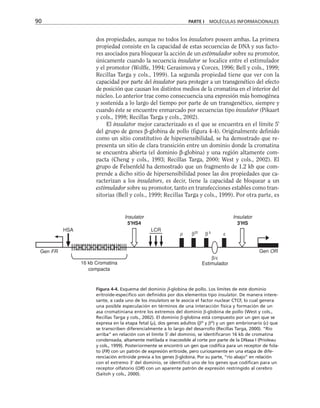
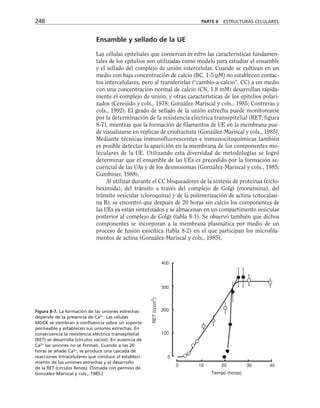
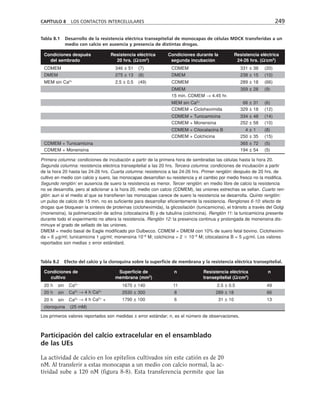
Figura 8-10. Efecto de distintas drogas sobre el desarrollo de la RET, durante un
cambio a calcio. La primera y segunda columnas representan los valores control
antes y después de 5 hrs. de transferencia a calcio respectivamente. Para facilitar
las comparaciones, el nivel de RET control adquirido con la transferencia a calcio
se marca con una línea discontinua. En la parte inferior se indican los inhibidores y
estimuladores empleados y en la porción superior las moléculas sobre las cuales ac-
túan. Prot G (proteína G): efecto de 14 ng/ml toxina pertussis y de 2 mM AlF3
; FLC
(fosfolipasa C): efecto de 110 µM de sulfato de neomicina y 2 µM de hormona liberado-
ra de tirotropina (HLT); PKC (proteína cinasa C): efecto de 50 µM 1-(5-isoquinolinil-
sulfonil)-2-metilpiperazina dihidrocloruro (H7) y 100 µg/ml de 1,2-dioctanoilglicerol
(diC8); CaM (calmodulina): efecto de 25 µM dihidrocloruro de trifluoperazina (TFPZ);
[AMPc]: efecto del dibutiril-AMPc (dB-AMPc), forskolina, e isobutil-metil-xantina
(IBMX). (Tomada con permiso de Balda y cols., 1991.)](https://image.slidesharecdn.com/biologia-celular-y-molecular-220613202029-80f4c21c/85/biologia-celular-y-molecular-pdf-292-320.jpg)






































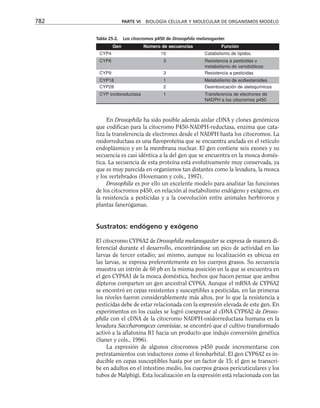
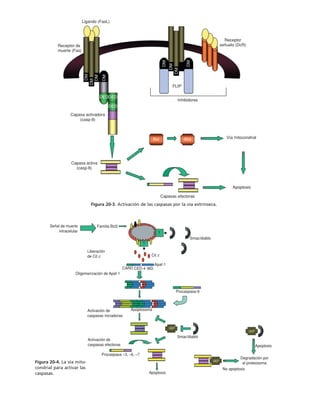
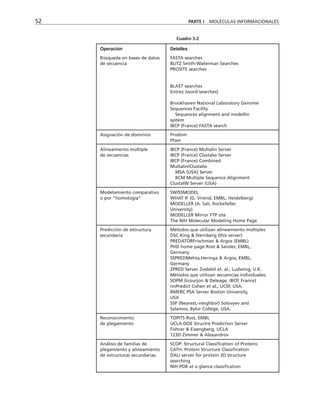
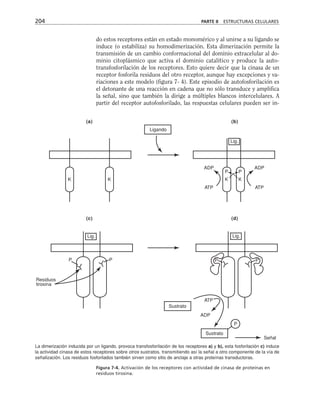
![290 PARTE II ESTRUCTURAS CELULARES
Microtúbulos* Microfilamentos* Filamentos intermedios*
MAPs Miosinas: contracción y Filagrina
desplazamiento (motores)
Factor TAU Sinemina
Epinemina
Filensina
Paranemina
Dinaína Proteínas
Nexina motoras
Cinesina]
Fimbrina: forma haces
Vellosina rompen filamentos
Gelsolina] y sellan sus extremos
Alfa actinina entrecruzadoras
Beta actinina]
Vinculina se asocian a placas
Talina ] de adhesión
Caldesmon
Espectrina: forma redes
Acumentina: sella extremos de
los filamentos
Tropomiosina estabilizan los
Troponina ] microfilamentos
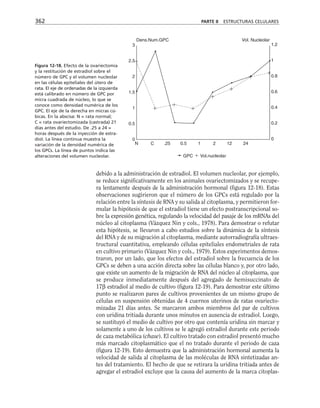
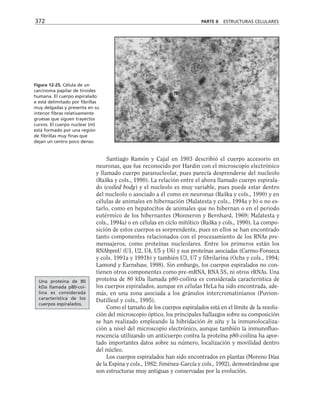
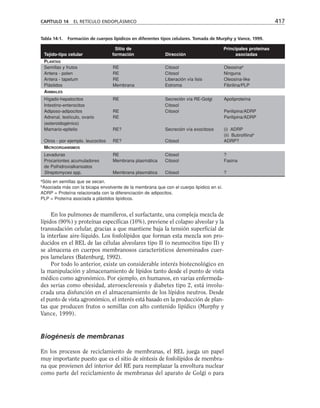
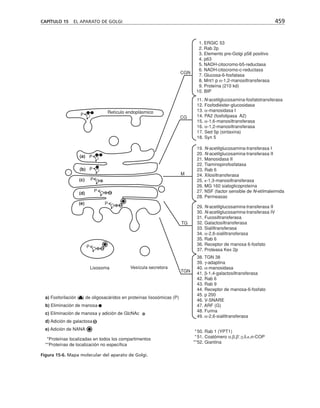
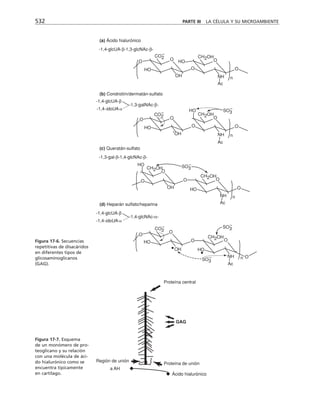
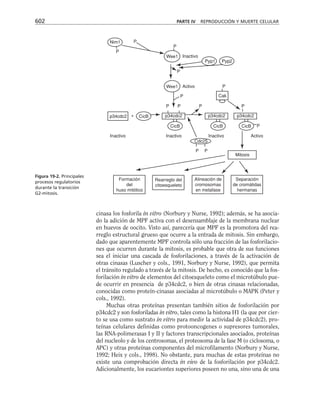
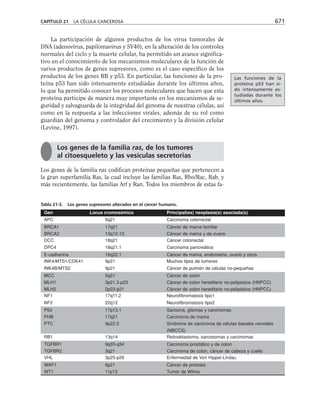
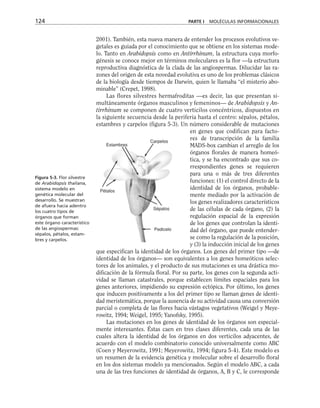
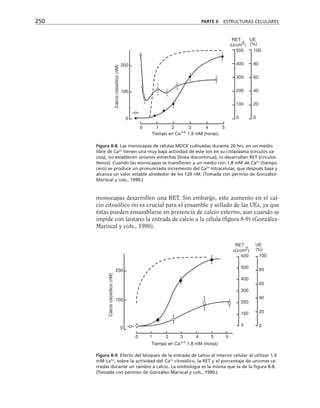
Tabla 9-1. Proteínas asociadas a elementos del citoesqueleto.
* Se mencionan sólo las mejor caracterizadas.
na soluble como a los filamentos de actina para formar haces y estructuras
más complejas (tabla 9-1).
Una gran proporción de la actina soluble está asociada a una proteí-
na llamada profilina, con peso molecular de 16,000 daltones. Este péptido
se une reversible y rápidamente a la actina G, retardando su polimeri-
zación en filamentos. La desoxirribonucleasa I se une también a actina
soluble, aunque hay algunas excepciones como es el caso de Entamoeba
histolytica y Naegleria. Otras proteínas que se unen a la actina, como
la filamina y la alfa-actinina hacen entrecruzamientos con los microfila-
mentos, produciendo redes. La vellosina y la gelsolina son proteínas que
rompen los filamentos de actina con actividad dependiente de calcio, y
sirven como centros de nucleación en el ensamble de los microfilamen-
tos. Caldesmon ayuda a formar haces de filamentos de actina. Algunas
proteínas se unen al extremo que crece de los filamentos durante la po-
limerización e impiden la adición de nuevos monómeros. Vinculina y
talina se localizan junto con la actina en las placas de adhesión que for-
man las células al adherirse a un sustrato. Algunos receptores membra-
nales se conectan con filamentos de actina para moverse en la superficie
celular.
La acción coordinada de todas estas proteínas regula las funciones, que,
en forma general, se refieren a funciones del citoesqueleto, por lo que su es-
tudio requiere de enfoques múltiples que permitan determinar las bases
moleculares de las estructuras para, de ahí, poder entender la función. En](https://image.slidesharecdn.com/biologia-celular-y-molecular-220613202029-80f4c21c/85/biologia-celular-y-molecular-pdf-331-320.jpg)