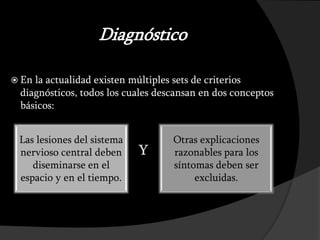
El documento resume información sobre la esclerosis múltiple (EM), incluyendo su historia, epidemiología, etiopatogenia, manifestaciones clínicas, diagnóstico y exámenes complementarios. Jean-Martin Charcot se le atribuye haber definido los primeros criterios diagnósticos para la EM. La EM es una enfermedad crónica del sistema nervioso central caracterizada por focos de inflamación y desmielinización que se diseminan en el espacio y el tiempo. Su diagnóstico se basa en hallazgos clínicos, de