Descargado 553 veces














































































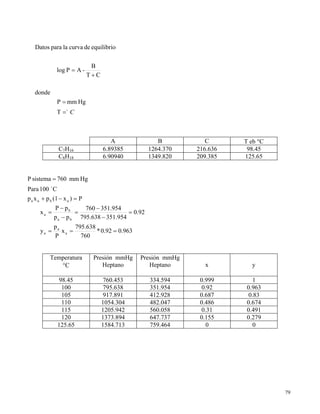
































































































































































Este documento describe el método de Mc Cabe-Thiele para diseñar columnas de destilación fraccionada. El método utiliza diagramas de equilibrio de presión-composición y temperatura-composición para determinar el número teórico de platos requeridos para separar los componentes de una mezcla binaria. También explica conceptos teóricos como equilibrio de fases, entalpía, presión de vapor, volatilidad relativa y leyes de Raoult y Trouton que son relevantes para la destilación fraccionada. Finalmente,















































