- Se presenta un caso de síndrome de Edwards diagnosticado ecográficamente desde la semana 18 de gestación y confirmado por cariotipo post natal. El recién nacido presentó varias malformaciones y falleció a las 48 horas de vida.
Caso clínico
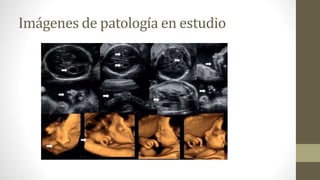
Paciente de19 años, nulípara, sin antecedentes gineco-obstétricos, se realiza ecografía obstétrica de
control en la semana 18 constatándose: feto único, vivo, situación pelviana, quiste de plexo coroideo,
labio paladar hendido y ausencia de mano en brazo izquierdo. Posteriormente se realiza ecografía
morfológica en semana 22 con los siguientes hallazgos: quiste de plexo coroideo bilateral, cisterna magna
aumentada, labio paladar hendido unilateral, micrognatia, oreja de implantación baja, mano en garra
derecha, ausencia de antebrazo izquierdo, sindactilia, comunicación interventricular, hidronefrosis, pie en
mecedora, placenta de inserción baja, arteria umbilical única. Los estudios posteriores y valoración con 3D
y 4D confirman los mismos hallazgos. Se plantea cariotipo por punción del cordón pero la paciente y
familiares no aceptan. Se procede a cesárea programada a las 37 semanas por situación transversal,
restricción del crecimiento intrauterino simétrico y placenta previa oclusiva lateral. Se obtiene un recién
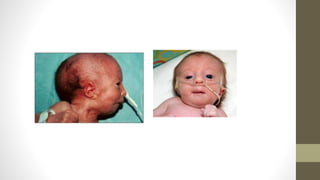
nacido vivo de 2100 gramos, sexo femenino, Apgar 6/7 y pasa a Unidad de terapia intensiva neonatal. Al
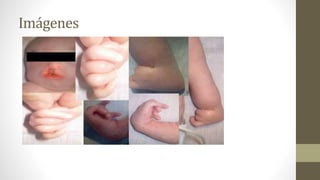
examen del recién nacido se constata el labio paladar hendido unilateral a derecha, micrognatia, mano
derecha en garra con superposición del 2° dedo sobre el 3° y del 5° sobre el 4° dedo, aplasia radial
izquierda, sindactilia izquierda, pie en mecedora unilateral derecha. Se realiza estudios de ecocardiografía
neonatal y ecoencéfalo constatándose los hallazgos descriptos prenatalmente. Se realiza cariotipo en
sangre periférica con resultado 47, XX+18. Se produce el óbito a las 48 hs post nacimiento.
3.
Resumen
• Se presentaun caso de síndrome de Edwards, por sospecha ecográfica desde la
semana 18 de gestación, con seguimiento ecográ- fico 2D, 3D y 4D, confirmado por
cariotipo post natal. Óbito 48 hs post nacimiento.
5.
Síntomas
• Puños cerrados.
•· Piernas cruzadas.
• · Pies con un fondo redondeado.
• · Bajo peso al nacer.
• · Orejas de implantación baja.
• · Uñas insuficientemente desarrolladas.
• · Cabeza y mandíbula pequeñas.
• · Forma inusual del pecho.
• · Retraso mental.
6.
¿Cómo se diagnostica?
•El síndrome de Edwards se puede diagnosticar durante el embarazo, cuando
mediante un examen, el médico observa un útero inusualmente grande y líquido
amniótico de más. Por otro lado, también puede diagnosticarse cuando el niño nace,
ya que puede que la placenta sea inusualmente pequeña. El examen físico del recién
nacido puede mostrar patrones inusuales de las huellas dactilares y las radiografías
pueden evidenciar un esternón corto. Otros signos que pueden indicar que el bebé
padece el síndrome de Edwards son: agujero, división o hendidura en el iris del ojo,
separación entre el lado izquierdo y derecho del músculo abdominal y hernia
umbilical o hernia inguinal. El bebé también puede mostrar signos de cardiopatía
congénita.
7.
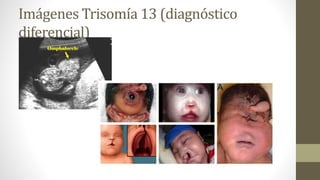
Diagnóstico diferencial
Trisomía 13Sx de Moessinger Sx de Edwards
• Holoprosencefalia
• Fisura de labio central
• Fisura palatina
• Frente en declive
• Microftalmia
• Hipotelorismo
• Puente nasal prominente
• Cuello corto
• Dextrocardia
• Polidactilia manos y pies
• movilidad fetal escasa,
• cordón umbilical corto,
• hipertelorismo,
• fisura palatina,
• contracturas articulares
múltiples
• hipoplasia pulmonar
• testes no descendidos
• pliegues de flexión palmo-
plantares hipoplásicos,
• Panículo adiposo y masa
muscular escasa al nacer
• microcefalia*, fontanelas
amplias,
• Defectos oculares
microftalmía, coloboma de
iris),
• fisuras palpebrales cortas
• orejas displásicas
• *micrognatia*,
• boca pequeña
• paladar ojival,
• labio/paladar hendido
• Puños cerrados
• testes no descendidos,
8.
Diagnóstico diferencial
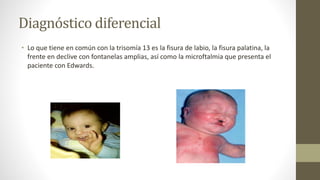
• Loque tiene en común con la trisomía 13 es la fisura de labio, la fisura palatina, la
frente en declive con fontanelas amplias, así como la microftalmia que presenta el
paciente con Edwards.
9.
Diagnóstico diferencial
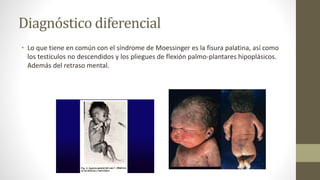
• Loque tiene en común con el síndrome de Moessinger es la fisura palatina, así como
los testículos no descendidos y los pliegues de flexión palmo-plantares hipoplásicos.
Además del retraso mental.
Presentación clínica
• Frecuenciaaproximada de 1:6000-8000 nacimientos
• 1 de cada 3.000 nacidos vivos y es tres veces más común en las niñas que en los
niños.
• Mayor frecuencia mujeres de edad avanzada por sobre los 35 años, aunque puede
presentarse en embarazos de mujeres jóvenes
• No existe evidencia respecto a que su prevalencia esté relacionada con razas o zonas
geográficas en particular.
• Mortalidad del 95% en el primer año de vida. El 5% suele vivir más tiempo (3% a los
5 años) y menos del 1% sobre los 5 años.
13.
Complicaciones
• Entre un90 y 95% de los afectados muere durante el primer año de vida (Bustillos-
Villalta y Quiñones-Campos, 2014).
• La supervivencia media se sitúa entre 2,5-70 días (Bustillos-Villalta y Quiñones-
Campos, 2014). Por lo tanto, son escasos y excepcionales los casos que alcanzan la
etapa adolescente (Simón-Bautista et al., 2008).
• De esta forma, las principales causas de fallecimiento son las cardiopatías
congénitas, las apneas y la neumonía (Pérez Aytés, 2000).
14.
Complicaciones
• Además, entreaquellos que superan los primeros años de vida, también se
presentan otro tipo de complicaciones médicas (Pérez Aytés, 2000):
• Problemas de alimentación.
• Escoliosis.
• Estreñimiento.
• Infecciones recurrentes (otitis, neumonía, etc.).
• Retraso psicomotor significativo.
15.
Factores de riesgo
•A pesar de que el síndrome de Edwards se produce de forma aislada en familias sin
antecedentes, se han identificado algunos factores que incrementan su probabilidad
de ocurrencia (Pérez Aytés, 2000):
• El riesgo de recurrencia en familias con otros casos es de 0.55%.
• Más probable cuando la madre tiene una edad más avanzada durante la gestación, a
partir de los 35 años la frecuencia se incrementa de forma progresiva.
16.
Estudios necesarios paradiagnóstico
• Por lo general, la presencia de marcadores ecográficos, anomalías anatómicas o las
pruebas bioquímicas en el suero materno suelen, suelen ofrecer indicadores fiables
de su presencia (Saldarriaga et al., 2016).
• Independientemente del momento de la exploración, para confirmar el diagnóstico
se extrae una muestra de ADN, y se realiza un cariotipo (imagen de la configuración
cromosómica) para confirmar las alteraciones en el par 18 (Trisomi 18 Foundation,
2016).
17.
Tratamiento
• Actualmente noexiste ningún tratamiento curativo para el síndrome de Edwards.
Además, la escasa supervivencia dificulta el diseño de intervenciones terapéuticas
específicas.
• A pesar de que no se conocen con exactitud los factores que contribuyen a la
supervivencia prolongada de los individuos que padecen síndrome de Edwards,
todas intervenciones médicas se orientan a paliar las complicaciones médicas
secundarias.
• De esta forma, lo más beneficioso es emplear un tratamiento rehabilitador integral
consistente en terapia física, cognitiva, ocupacional, entre otras.
Comentarios
El síndrome deEdwards es una de las anomalías cromosómicas con altas tasas de
mortalidad neonatal. Por lo que ante una sospecha por hallazgos en ecografía 3D y 4D,
como en el ejemplo del caso previsto, se debe plantear una confirmación genética y
tener el manejo por medio de un equipo multidisciplinario con experiencia, ya sea
desde el obstetra, neonatólogo, genetista, hasta el psicólogo para afrontar cualquier
noticia grave respecto al neonato. La ayuda de la ecografía es indispensable para una
evaluación previa. Esta patología resulta tener mayor incidencia en niñas, teniendo una
de cada 3.000 como probabilidad y suele ocurrir más seguido en embarazos por
encima de los 35 años de edad, aunque como vimos en el caso previo, suele suceder
también en embarazos a temprana edad, en este caso la mujer tenía 19 años.
23.
• El impactoque tiene esta patología sobre la sociedad, suele tener un cambio de
perspectiva en cuanto al cuidado de la madre en el embarazo, y a cuidarse de
cualquier teratógeno que pueda conllevar a este síndrome al recién nacido.
• Igualmente en la familia de la paciente puede tener un impacto muy fuerte, por lo
que es necesario también la ayuda psicológica no solo en la madre embarazada, sino
también en la familia para afrontar la gran noticia de que el pequeño va a nacer con
este conjunto de malformaciones. Aquí reside la gran importancia de la
interdependencia del área clínica, recalcando que no sólo se trata de los médicos y
enfermeros, sino también de apoyo para tomar decisiones tanto éticas como
morales.
24.
Propuesta
• Como propuesta,podríamos implementar campañas para el cuidado de la
embarazada ante cualquier sustancia teratógena, así como el chequeo por medio de
ecografía antes de la semana 15 para saber en que condiciones se encuentra el
producto a nacer. Mientras que después de nacido no se puede hacer mucho para
mejorar la calidad de vida del neonato, puesto que este suele no superar los 15 días
de vida.
25.
Bibliografía
• 1. GardnerRJM, Sutherland GR. Down syndrome, other full aneuploidies and polyploidy (En:
Chromosome abnormalities and genetic counselling. Gardner RJM, Sutherland GR eds.) Oxford
university Press Inc., New York 1996, pp 252
• 2. Martínez-Frías ML. Tabla 3.17: Síndromes cromosómicos (En: Defectos congénitos en España:
Diez años de vigilancia epidemiológica. Dirección General de planificación Sanitaria, Ministerio de
Sanidad y Consumo ed.) Secretaría General Técnica, Publicaciones, Madrid 1989, pp. 41
• 3. Jones KL. Trisomy 18 syndrome (En "Smith´s Recognizable Patterns of Human Malformation.
Jones KL ed.) W.B. Saunders Co, Philadelphia1997, pp 16-17
• 4. Hodes ME, Cole J, Palmer CG, Reed T. Clinical experience with trisomies 18 and 13. J Med Genet
1978;15:48-60
• 5. Boghosian-Sell L, Mewar R, Harrison W, et al. Molecular mapping of the Edwards syndrome to
two noncontiguous regions on chromosome 18. Am J Med Hum Genet 1994;55:476-83
• 6. Marion RW, Chitayat D, Hutcheon G, et al. Trisomy 18 score: A rapid, reliable diagnostic test for
trisomy 18. J Pediatr 1988;113:45-8
• 7. Baty B, Blackburn B, Carey J. Natural history of trisomy 18 and 13: I. Growth, physical
assessment, medical histories, survival and recurrence risk. Am J Med Genet 49:175-188, 1994
• 8. Baty B, Jorde L, Blackburn B, Carey J. Natural history of trisomy 18 and 13: II. Psychomotor
development. Am J Med Genet 49:189-194, 1994.
26.
Referencias (caso clínico)
•1. Cafici D, Mejides A, Sepulveda W. Ultrasonografía en obstetricia y diagnóstico
prenatal. Buenos Aires: Journal, 2007. p. 561-562
• 2. Pérez Aytés A. Trisomía 18 (síndrome de Edwards). Protoc diagn ter pediatr.
2010;1:96-100
• 3. Rumack C, Wilson S, Charboneau J. Ecografia obstétrica y fetal. Madrid. 2000 p.
305-325.
• 4. Mauad F, Pinheiro L. Ultrasonografia na practica obstétrica. Rio de Janeiro.
Revinter. 2006. p 137-139
• 5. Melo N, Fonseca E. Medicina fetal. Rio de Janeiro: Elsevier, 2012. p 49-58
• 6. Drose J. Ecocardiografia fetal. 2° edición. Venezuela: Amolca. 2011. p 105-116
• 7. Gallo M, Santiago J, Ramos Corpas D. Ecogrfia fetal.Venezuela: Amolca. 2010.
• 8. Gallo M, Sanchez R. Ecografia fetal de semana 18-22 de Embarazo. Venezuela:
Amolca. 2014.