Descargado 171 veces



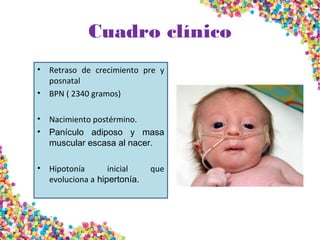
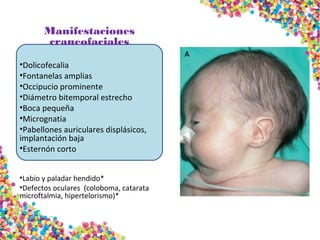
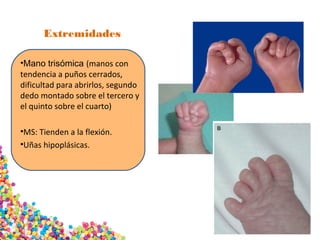

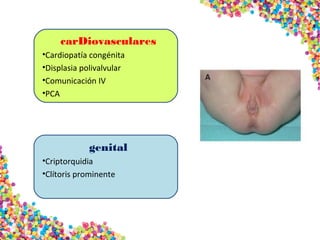
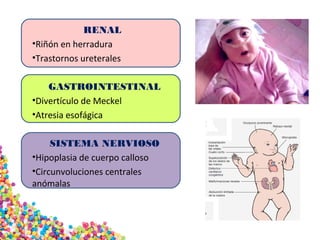
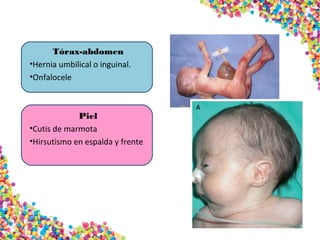
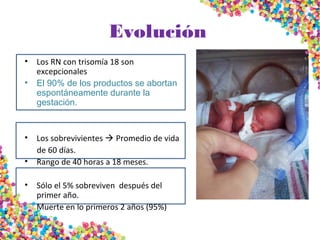

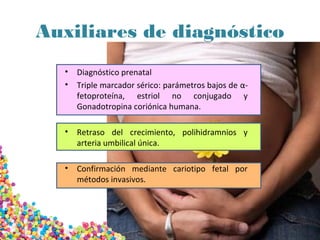


La trisomía 18 o síndrome de Edwards se caracteriza por la presencia de una copia adicional del cromosoma 18 y causa anomalías en diversos órganos. Los bebés con este síndrome generalmente presentan retraso en el crecimiento, defectos craneofaciales y cardíacos, y solo el 5% sobreviven más de un año, con una esperanza de vida promedio de dos meses. El diagnóstico se confirma mediante el análisis de cariotipo fetal.




























































































