UNIVERSIDAD PARA ELBIENESTAR
‘’BENITO JUÁREZ GARCÍA’’.
SANTIAGO YOLOMECATL
LICENCIATURA EN MEDICINA INTEGRAL Y SALUD COMUNITARIA.
MEDICINA EN CAMPOS CLÍNICOS Y COMUNITARIOS III:
REUMATOLOGIA
• Barragán Flores Karen
• Cruz Hernández Daniela
• Cruz Velasco Fabiola Gpe.
• López Jiménez Yuridia
• Mendoza Barrios Heidy Lizbeth
• Pacheco García Joanna Brigitte
• Pérez Hernández Ana Gabriela
• Ramos Reyes Diana Miroslava
• Ramos Reyes Jairo Edu
• Sandoval Cruz Kimberly
INTEGRANTES:
CATEDRATICA: ENF. MARIBEL LILIAN GALICIA TAPIA
2.
“
Grupo heterogéneo deprocesos que reconocen como sustrato
patológico de la presencia de inflamación en los vasos
sanguíneos, pudiendo asociar necrosis en la pared vascular
2
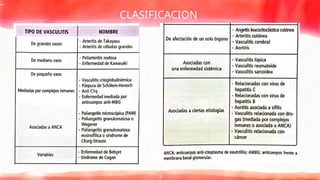
DEFINICIÓN
Etiopatogenia
▸ Para lamayoría de los síndromes vasculiticos
la etiología es desconocida, aunque parecen
mediadas por mecanismos inmunológicos.
▸ El depósito de inmunocomplejos en la pared
vascular activaría los factores del
complemento y desencadenaría una reacción
inflamatoria: vasculitis de mediano vaso y
vasculitis de pequeño vaso ANCA negativas.
5.
▸ La presenciade
anticuerpos dirigidos
frente al citoplasma
de los neutrófilos
(ANCA) es un
hallazgo frecuente en
algunas vasculitis:
vasculitis de pequeño
vaso ANCA positivas.
▸ La hipersensibilidad
retardada e inmunidad
celular también podrían estar
implicadas como responsables
de los granulomas que
aparecen en algunas formas
de vasculitis: vasculitis de gran
vaso y granulomatosis con
poliangeítis y eosinofílica.
6.
diagnostico
▸ Por sussimilitudes clínicas se
engloban como vasculitis necrotizantes
sistémicas: la panarteritis nodosa
clásica (PAN), la poliangeítis
microscópica, la granulomatosis con
poliangeítis y la granulomatosis
eosinofílica con poliangeítis.
▸ Muestran una clínica donde
coexiste un síndrome
constitucional con astenia,
anorexia, febrícula y pérdida de
peso junto con afectación
multisistémica.
▸ Se realiza mediante una clínica compatible y una
confirmación histológica. Sin embargo, en algunos
cuadros, el diagnóstico es clínico (Kawasaki) o por
técnicas de imagen (Takayasu).
7.
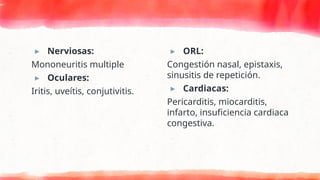
Sospecha diagnostica
▸ Síndromeconstitucional:
Fiebre, astenia, malestar,
artromialgias..
▸ Cutáneas:
Purpura palpable: Suele comenzar en
zonas declives, nódulos subcutáneos,
urticaria crónica, livero reticularis,
ulceras, telangiectasias del lecho
ungueal, infarto o gangrena digital.
8.
▸ Renales:
Es lamas
característico y
frecuente de la PAN.
Puede cursar con
glomerulonefritis,
proteinuria,
hematuria,
insuficiencia renal,
HTA.
▸ Pulmonares:
Hemoptisis, tos,
disnea, crisis
asmáticas.,
alteraciones
radiográficas
(nódulos, infiltrados).
▸ Digestivas
Melenas, dolor
abdominal, nauseas,
vómitos, infarto y
perforación intestinal.
Pruebas de laboratorio
▸Biopsia.
▸ Estudios de sangre: reactantes de fase aguda,
ANCA, factor reumatoide.
▸ Estudios de imagen vascular.
▸ AngioRM
▸ AngioTC
▸ Angiografía
11.
Tratamiento
▸ FASE MÁSINTENSA (DE INDUCCIÓN DE LA REMISIÓN): Se
administran corticoides en dosis altas (1 mg/kg/día durante el primer
mes). Se suele añadir al tratamiento Ciclofosfamida si hay afectación
de órganos vitales, lo que es frecuente.
▸ La Ciclofosfamida se puede administrar por vía oral o intravenosa.
La administración en bolos intravenosos permite alcanzar una dosis
acumulada inferior y minimizar los efectos secundarios a largo plazo.
▸ Los principales efectos secundarios son pancitopenia, mielodisplasia,
aumento de la incidencia de infecciones, cistitis hemorrágica, cáncer
vesical o toxicidad gonadal.
12.
TRATAMIENTO DE MANTENIMIENTO:
▸Se suele llevar a cabo con otro inmunosupresor
de perfil más seguro, como Metotrexato,
Azatioprina o Micofenolato de mofetilo.
▸ En las ANCA positivas Rituximab, tanto en
inducción como en mantenimiento.
▸ También en las ANCA positivas la
Plasmaféresis se puede considerar en algunos
casos, especialmente en aquellos en los que
haya afectación renal grave.
13.
Diagnostico Diferencial
▸ Infecciones:Algunas infecciones, como la endocarditis bacteriana, la
enfermedad de Lyme, la sífilis y la hepatitis viral, pueden causar síntomas
que imitan los de la vasculitis.
▸ Trastornos autoinmunes: Enfermedades autoinmunes como el lupus
eritematoso sistémico (LES), la artritis reumatoide y la esclerodermia pueden
presentar síntomas similares a los de la vasculitis y pueden coexistir con
vasculitis o ser diagnosticadas erróneamente como vasculitis.
▸ Alergias: Reacciones alérgicas graves, como la anafilaxia o la vasculitis
alérgica inducida por fármacos, pueden causar síntomas de vasculitis.
▸ Trastornos trombóticos: Los trastornos de la coagulación, como el
síndrome antifosfolípido, la trombosis venosa profunda y la trombocitopenia
trombótica púrpura (TTP), pueden presentar síntomas similares a los de la
vasculitis debido al daño vascular.
14.
▸ Enfermedades
metabólicas:
Condiciones como
ladiabetes
mellitus y la
amiloidosis
pueden afectar
los vasos
sanguíneos y
causar síntomas
similares a los de
la vasculitis.
▸ Cáncer: Algunos
tipos de cáncer,
especialmente el
linfoma y el
carcinoma de
células renales,
pueden presentar
síntomas de
vasculitis como
parte de su
presentación
clínica.
▸ Reacciones adversas
a medicamentos:
Algunos
medicamentos pueden
causar reacciones
adversas que imiten
los síntomas de la
vasculitis, como la
purpura por IgA
inducida por fármacos
o la vasculitis inducida
por antiinflamatorios
no esteroides (AINEs).
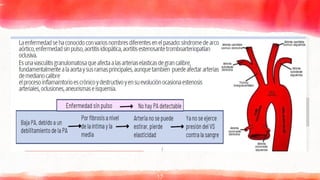
▸ La arteritisde células
gigantes es una inflamación
de la capa que recubre las
arterias.
▸ Compromete vasos de
mediano y gran calibre,
afectando a las ramas
aórticas, con predilección
por las ramas de la
carótida, en especial la
arteria temporal.
▸ Prevalencia en > 55 años,
mayoritariamente mujeres
50% de los casos se asocian a
polimialgia reumática.
23.
Manifestaciones clínicas
▸ Laedad media: 70 años
▸ síntomas inespecíficos: pérdida de peso, astenia, anorexia,
artralgias, febrícula y malestar general.
▸ Los síntomas más específicos son: Cefalea (65%) no habitual
y refractaria a los analgésicos habituales, engrosamiento de
la arteria afectada, nódulos subcutáneos o ausencia de pulso
temporal; ocular (25-50%) oclusión de diferentes arterias
oculares u orbitarias: perdida de la visión.
▸ Oclusión permanente: neuritis óptica isquémica
▸ Claudicación mandibular: dolor al masticar o en la lengua.
24.
Diagnóstico
▸ Se debesospechar ante el cuadro clínico
compatible, pero el diagnóstico
definitivo se debe realizar por biopsia de
la arteria afectada, se debe llevar a cabo
lo antes posible, ya que el tratamiento
con corticoides puede enmascarar
algunos de los hallazgos histológicos.
▸ Ecografía de las arterias temporales,
que puede mostrar hallazgos muy
sugestivos como el halo hipoecoico,
alrededor de la arteria afectada o el
engrosamiento de la pared con
disminución del flujo.
25.
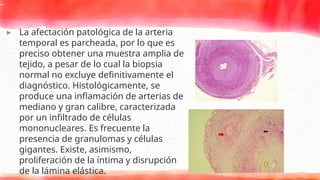
▸ La afectaciónpatológica de la arteria
temporal es parcheada, por lo que es
preciso obtener una muestra amplia de
tejido, a pesar de lo cual la biopsia
normal no excluye definitivamente el
diagnóstico. Histológicamente, se
produce una inflamación de arterias de
mediano y gran calibre, caracterizada
por un infiltrado de células
mononucleares. Es frecuente la
presencia de granulomas y células
gigantes. Existe, asimismo,
proliferación de la íntima y disrupción
de la lámina elástica.
26.
TRATAMIENTO
▸ Es concorticoides, no sólo son
eficaces en el alivio sintomático,
sino también en la prevención
de las complicaciones oculares.
▸ La dosis empleada es de 1
mg/kg/día durante las primeras
semanas.
▸ Afectación ocular: realizar
inicialmente tres bolos de 1
gramo de metilprednisolona
por 3 días.
▸ Corticoides mg/kg/día + calcio y
vitamina D + bifosfonatos
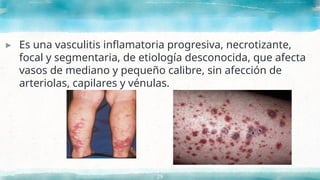
▸ Es unavasculitis inflamatoria progresiva, necrotizante,
focal y segmentaria, de etiología desconocida, que afecta
vasos de mediano y pequeño calibre, sin afección de
arteriolas, capilares y vénulas.
29
Diagnostico:
31
• Biopsias.
• USGDoppler.
• Resonancia magnética.
Tratamiento:
Corticoides: 1mg/kg/día.
Inmunosupresores, en caso de afección severa.
Ciclofosfamida IV.
Azatioprina o metotrexato.
CONCEPTO:
Enfermedad multisistémica, convasculitis de pequeños y medianos
vasos propia de lactantes y niños pequeños (80% de casos en
menores de 5 años).
33
ETIOLOGÍA:
Desconocida
Etiología infecciosa,
presumiblemente por c. pneumoniae
FASES:
- Aguda febril (1-2 sem)
- Subaguda
- Convalecencia
POLIANGEITIS MICROSCOPICA
▸ Característicashistológicas y clínicas similares a la
PAN, pero afecta principalmente vasos de pequeño
calibre especialmente capilares y vénulas
40.
Diferencias con laPAN
▸ No aparecen
microaneurismas,
no forma
granulomas
▸ Afección
pulmonar
▸ Glomerulonefritis
▸ TRATAMIENTO
▸ Corticoesteroides
▸ Inmunosupresore
s: Ciclofosfamida
o Rituximab
▸ La plasmaferesis
en afeccion renal
grave
41.
GRANULOMATOSIS CON POLIANGEITIS
▸Vasculitis granulomatosa
necrozante que afecta vías
respiratorias superiores e
inferiores, glomérulos renales
▸ Etiologia desconocida
Frecuente desde los 40 años
Granulomatosis de Wegener
42.
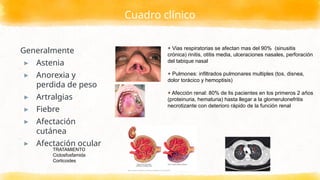
Cuadro clínico
Generalmente
▸ Astenia
▸Anorexia y
perdida de peso
▸ Artralgias
▸ Fiebre
▸ Afectación
cutánea
▸ Afectación ocular
+ Vias respiratorias se afectan mas del 90% (sinusitis
crónica) rinitis, otitis media, ulceraciones nasales, perforación
del tabique nasal
+ Pulmones: infiltrados pulmonares multiples (tos, disnea,
dolor torácico y hemoptisis)
+ Afección renal: 80% de lis pacientes en los primeros 2 años
(proteinuria, hematuria) hasta llegar a la glomerulonefritis
necrotizante con deterioro rápido de la función renal
TRATAMIENTO
Ciclosfosfamida
Corticoides
43.
Vasculitis granulomatosa deChurg-Strauss (poliangeítis
granulomatosa eosinofilica)
▸ Necrotizante sistémica
granulomatosa.
▸ Caracterizada por afección
pulmonar, historia de
asma grave y atopia.
▸ Etiología desconocida.
▸ Mas frecuentes en varones
Anatomía patológica
Vasculitis necrotizante de arterias musculares
de pequeño y mediano calibre.
Infiltración tisular por eosinofilos.
Granulomas vasculares y extravasculares.
44.
Cuadro clínico
• Manifestacionesgenerales del proceso
(fiebre, adelgazamiento, malestar general)
• Predominio de asma en fase inicial
• Puede encontrarse afectación crónica de
senos paranasales
• Afectación pulmonar: infiltrados pulmonares
migratorios, no cavitados y bilaterales.
• Miocardiopatía restrictiva
• Nódulos subcutáneos, dolor abdominal (de
diversas causas), mononeuritis múltiple,
artritis no erosiva y glomerulonefritis.
Diagnóstico
Sospecha clínica y de laboratorio (p-ANCA, con
confirmación histológica)
Criterios de clasificación
• Asma
• Eosinofilia >10 % del recuento leucocitario
• Mononeuropatías, mononeuretis múltiple o polineuropatía
• Infiltrados pulmonares fugaces no cavitados
• Historia de sinusitis aguda o crónica o veladura
radiológica de los senos paranasales
• Presencia de infiltración eosinofila extravascular
45.
Tratamiento
Fase de remisiónFase de mantenimiento
Prednisona 1 mg/kg/día, asociado a un inmunosuprosor:
• Si existe daño orgánico o amenaza vital se usa
ciclofosfamida o rituximab.
• Si no existe daño orgánico o amenaza vital se utiliza
metotrexato o micofenolato.
se disminuye progresivamente la dosis de esteroides y
asociamos fármacos: azatioprina, rituximab, metotrexato o
micofenolato (deben durar menos de 24 meses)