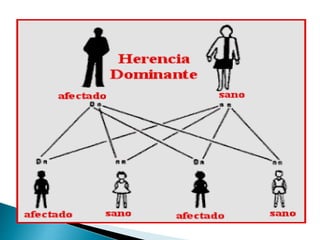
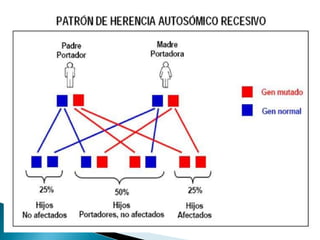
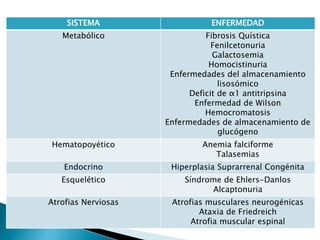
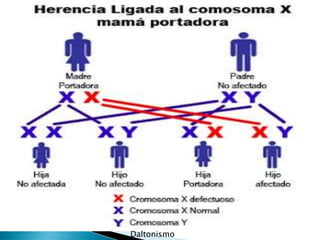
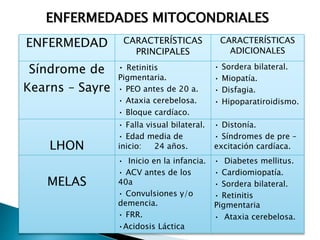
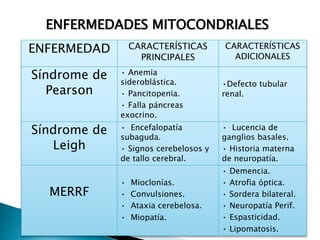
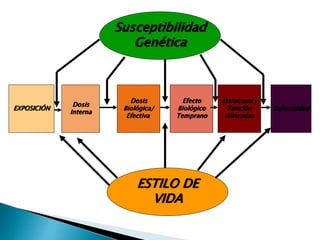
Este documento trata sobre genética clínica. Explica conceptos básicos como gen, locus, alelos y tipos de herencia como dominante, recesiva y ligada al sexo. Describe varios síndromes y trastornos genéticos, sus patrones de herencia y características. También cubre temas como herencia mitocondrial, multifactorial y poligénica.