Descargar para leer sin conexión











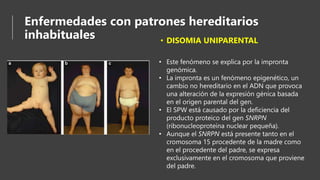










Este documento describe diferentes patrones de herencia genética como las enfermedades autosómicas dominantes y recesivas, las enfermedades ligadas al cromosoma X, las enfermedades multifactoriales, y patrones hereditarios inusuales como la herencia mitocondrial, la disomía uniparental y la expansión de trinucleótidos repetidos. También discute los teratógenos y la importancia de la valoración genética para identificar enfermedades genéticas.























































































