Descargado 108 veces









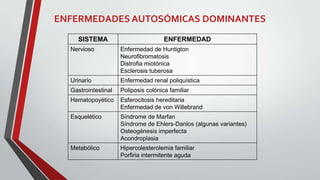
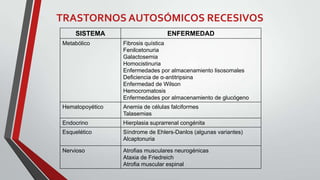

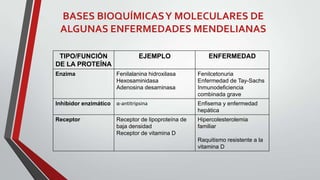
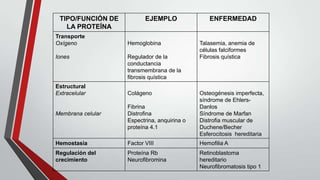












Este documento describe diferentes tipos de trastornos hereditarios, incluyendo trastornos monogénicos autosómicos dominantes y recesivos, ligados al sexo, y multifactoriales. Explica las bases moleculares de algunas enfermedades como la fibrosis quística, la fenilcetonuria, la talasemia y la enfermedad de Tay-Sachs. También describe síndromes como el síndrome de Marfan, la hipercolesterolemia familiar, la enfermedad de Gaucher y los síndromes de Down, Klinefelter y Turner















































