Descargado 11 veces








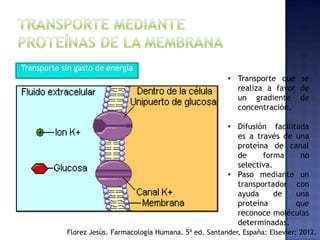









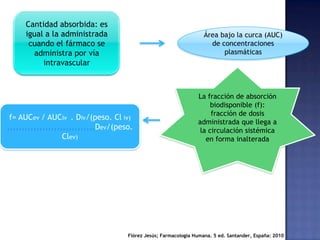













El documento trata sobre los procesos de absorción de fármacos en el organismo. Explica que la absorción incluye la liberación del fármaco de su forma farmacéutica, la disolución y la absorción propiamente dicha. También describe factores que afectan la cinética de absorción como la velocidad, cantidad absorbida y vías de administración de fármacos.























































![CLASE 1 FARMACOLOGIA 2023 (1) [Autoguardado].ppt](https://cdn.slidesharecdn.com/ss_thumbnails/clase1farmacologia20231autoguardado-250521212444-ba122e2e-thumbnail.jpg?width=640&height=640&fit=bounds)







