El documento describe diferentes formas farmacéuticas y vías de administración de medicamentos. Cubre temas como formas sólidas, líquidas y semisólidas; medicamentos de liberación controlada; y vías de administración como oral, tópica, sistémica e inhalatoria. También explica conceptos como absorción, distribución, biotransformación y eliminación de fármacos en el organismo.

































![Importancia clínica de la unión F-P 1.- [ F ] libre actividad farmacológica y clearance renal 2.- Desplazamiento competitivo a.- entre drogas b.- por substancias endógenas ( bilirrubina, ác. Grasos ) Efecto farmacológico de la droga desplazada 3.- En hipoalbuminemias ( falla hepática, síndrome nefrótico ) [ F ] libre a cualquier dosis](https://image.slidesharecdn.com/fcocintica-1216509201800025-8/85/FcocineTica-34-320.jpg)











![Excreción v/s Eliminación No son sinónimos La excreción es parte de la eliminación. Consiste en la movilización de F o metabolitos al exterior (disminución de [F]p) . Es un proceso irreversible. La eliminación también provoca disminución de [F]p. Existe reversibilidad. El F de redistribuye.](https://image.slidesharecdn.com/fcocintica-1216509201800025-8/85/FcocineTica-46-320.jpg)











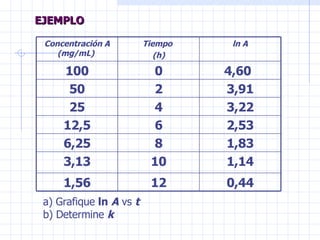
























































![CLASE 1 FARMACOLOGIA 2023 (1) [Autoguardado].ppt](https://cdn.slidesharecdn.com/ss_thumbnails/clase1farmacologia20231autoguardado-250521212444-ba122e2e-thumbnail.jpg?width=640&height=640&fit=bounds)



































![[NALL OCR] ESTULIN, Daniel (2007). Los señores de las sombras. Bogotá: Editor...](https://cdn.slidesharecdn.com/ss_thumbnails/nallocrestulindaniel2007-260104173047-32e96353-thumbnail.jpg?width=640&height=640&fit=bounds)
