ciencia y tegnologia acuativa para la conservacion
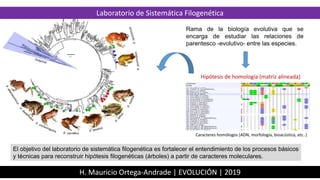
1. Laboratorio de Sistemática Filogenética
Rama de la biología evolutiva que se
encarga de estudiar las relaciones de
parentesco -evolutivo- entre las especies.
Hipótesis de homología (matriz alineada)
Caracteres homólogos (ADN, morfología, bioacústica, etc..)
El objetivo del laboratorio de sistemática filogenética es fortalecer el entendimiento de los procesos básicos
y técnicas para reconstruir hipótesis filogenéticas (árboles) a partir de caracteres moleculares.
H. Mauricio Ortega-Andrade | EVOLUCIÓN | 2019
2. Materiales y métodos |Práctica 1|
Revisión y edición de
secuencias (.ab1)
Alineamiento de
secuencias (.nex)
Modelo evolutivo de
sustitución de nucleótidos
Reconstrucción hipótesis
filogenética
Edición de árboles
http://www.geneious.com/
http://www.mbio.ncsu.edu/BioEdit/bioedit.html
http://phylemon.bioinfo.cipf.es/ev
olutionary.html
http://mrbayes.sourceforge.net/
http://jmodeltest.org/login http://www.robertlanfe
ar.com/partitionfinder/
https://www.phylo.org/portal2/login!input.action
https://code.google.com/arc
hive/p/garli/downloads
http://tree.bio.ed.ac.uk/software/figtree/
3.
4. Práctica 1 | Alineación de secuencias
Secuencia “limpia”
Secuencia con “ruido”
Criterio Técnico
5. Espacios
Espacios artificiales sin
codificación
Espacios artificiales con codificación
(N)
Espacios identificados como nucleótidos
(G)
No modificar, pues no
hay mala lectura en la
secuencia
Modificar, pues hay mala lectura
en la secuencia
Modificar y editar (borrar) nucleótido
identificado erróneamente
Situación
Decisión
10. 1. Abrir Geneious y crear una carpeta llamada “Lab1”
2. Cargar las secuencias almacenadas en la carpeta “Secuencias” (GoogleDrive)
3. Visualizar independientemente las secuencias del especímen QCAZ19180 (Forward |SarL| y Reverse |SbrH)
4. Realizar un consenso (assembly/contig) con el par de secuencias.
5. Editarlas de acuerdo criterio técnico, en función del nucleótido identificado en cada posición de la secuencia.
6. Realizar la edición y consenso para todas las secuencias del ejercicio.
Práctica 1 | Edición de secuencias y consenso (30 minutos)
https://seqcore.brcf.med.umich.edu/sites/default/files/html/interpret.html
11. 16S rRNA Secondary Structure
Loop
Tronco
Entre las regiones conservadas, el gen 16s RNA tiene alrededor
de 9 regiones hipervariables.
Para estudios de organismos cercanamente relacionados (a nivel
poblacional, p.ej.), las regions hipervariables son muy útiles para
diferenciar haplotipos.
16s RNA.
Para estudios filogenéticos a nivel de
especies, es preferible trabajar con las
regiones conservadas para construir
hípótesis de homología.
12. Práctica 1 | Alineamiento (30 minutos)
1. Con las secuencias consenso validadas, construya una matriz alineada (algoritmo Geniuous aligment)
2. Revise el resultado de la matriz alineada.
3. Editarla para que todas las secuencias formen una hipótesis de homología, es decir una matriz alineada.
4. Guarde una copia de la matriz alineada, con el nombre “16S_alineada”.
5. Cargue en Geneious la matriz llamada “16s_extraction.nex” de la carpeta “Matrices” y alinéala con la matriz
“16S_alineada”, y grábelo en un archivo nuevo, llamado “16S_sinHV” que significa, “16s sin hipervariables”.
14. Anotaciones en matrices: Excluir regiones hipervariables y extremos de las secuencias
1. Abra la matriz “16S_sinHV” en Mesquite.
2. Sobre este archivo, identifique, seleccione y anote como “Excluido” los extremos de las secuencies y las regiones
hipervariables.
3. De manera opcional, dependiendo del programa de análisis (BEAST, p. ej.), borre las áreas hipervariables.
Anotar caracteres seleccionados
desde el panel “Character List”
15. Práctica 1 | Matrices concatenadas de distintos genes (60 minutos)
Matrices concatenadas de varios genes
Master block
Archivo con los nombres
de todas las secuencias
a concatenar entre
genes
Matrices individuales
Matrices alineadas de
genes individuales (16s,
COI, RAG, etc)
Matriz concatenada
Matriz que concatena al
Master Block con las
matrices individuales
+ =
16. Práctica 1 | Matrices concatenadas de distintos genes (60 minutos)
1. New Project
2. New ->New Taxa Block -> Asigno el nombre “Master”, con
tres taxa asociados.
3. Incluir en el proyecto los archivos “16s.nex”, “COI.nex” y
“RAG.nex” que están en la carpeta “Matrices”.
MASTER BLOCK
17. 1. Renombrar los Taxa Block y
Character Matrix de acuerdo a
cada gen.
2. En menú principal voy a “Taxa
& Trees” -> New Association ->
Selecciono Master ->
selecciono la matriz con el
mayor número de especies, p.
ej. 16s -> Asigno el nombre a la
asociación “Master&16s”
¿Cuál de las matrices individuales tiene el mayor número de secuencias?
18. 1. Borro las tres primeras especies, si es que quedan vacías.
1. Selecciono todos los nombres de la lista de la derecha del menú
2. De los títulos de la columna 16s presiono “el triángulo negro invertido” en la columna 16s -> Create new taxa
in Master Block from Selected” -> aceptar.
19. 1. En menú principal voy a “Taxa & Trees” -> New Association ->
Selecciono Master -> ahora selecciono la matriz con el segundo lugar
en número de especies, p. ej. COI -> Asigno el nombre a la asociación
“Master&COI”.
Matriz que concatena varios genes
20. 2. Selecciono todos los nombres de la lista de la derecha del menú.
3. De los títulos de la columna COI presiono “el triángulo negro invertido” en la columna COI -> Auto assign
matches” / ignore White-spaces/ Ignore Cases / Matched if a number in both names are the same /
mínimum number of digits in number (5) -> aceptar.
OK!
21. Así debe verse el archivo master, con tres genes concatenados:
1. Ahora edito los nombres de las terminales o taxa en el Block del Master, desde el archivo Excel de la base
de datos con el siguiente formato: #Museo_Localidad_spp. Esto es necesario para estandarizar los
nombres entre las matrices (opcional).
2. Grabo el archivo como “MasterMatrixAssociated”, el cual contiene las asociaciones entre las bases de
datos y el Master.
23. Formato NEXUS de una matriz concatenada |Wordpad|
Encabezado
Especies grp. externo
Comando de “Fin de la matriz”
“Bug” en el archivo por defecto (borrarlo)
Detalles de las particiones del archivo
Instrucciones para MrBayes
24. Codificar en la matriz codones de Genes Codificantes
1. Cargar el archivo “MrBayes_concat.nex” y
renombrar el “Taxa block” y “carácter matrix”
como “MrBayes_codon”.
2. Matrix Genetic Code Selecciono regiones de
los genes en la matriz.
3. Asigno nombres de genes a regiones (16s, COI,
RAG).
25. 1. Selecciono la región de un gen codificante (p.ej. COI)
2. Codon position Set codón position Minimize Stop Codons Asigno nombres
de genes a regiones (16s, COI, RAG).
3. Realizar el mismo procedimiento con el gen codificante nuclear (RAG1) Codones identificados
26. 1. Exportar la matriz como archivo NEXUS para MrBayes, con el nombre MrBayes_codón.nex
2. Note que ahora ya trae las posiciones de los genes y los codones.
3. Crear una carpeta que se llame “Matrices individuales” y Exportar la matriz MrBayes_codón.nex como
Particiones separadas en archivos NEXUS.
Matriz codificada para codones Matrices de genes individuales
27. ¿Cuántas especies forman el grupo externo? ¿Cuántas terminales tiene la matriz?
¿Cuántos caracteres? ¿Cuántos genes codificantes?
30. NOTA: Es necesario tener instalado el programa Python
1. COPIAR la carpeta “PartitionFinderV1.1.1_Windows” en el disco C:, y RENOMBRARLA como
“PartitionFinder”.
1) En la ruta C:PartitionFinderexamples crear una carpeta llamada “CLASE” y pegar los dos archivos .py,
que contiene la matriz y el .cfg, que contiene las especificaciones del análisis.
31. 1. Abrir la consola de Windows, click derecho en símbolo
WINDOWS, “All Programs” -> “Accessories” -> “Command
Prompt”.
2. En la consola se debe describir tres comandos en la misma
línea ejecutable: “Python”, “la ruta donde está el archivos de
PartititionFinder.py”, la “carpeta” donde se encuentran los
archivos .phy y .cfg. Estos tres componentes deben estar en
la misma línea de comando de la consola, así:
a. Tipear “python“ seguido de un espacio.
b. Arrastrar a la consola el archivo “PartitionFinder.py”
(que está en la carpeta de descomprimido de
PartitionFinder). La ruta hacia ‘PartitionFinder.py’ se
cargará automáticamente a la consola.
c. Tipea otro espacio, y,
d. Arrastra a la consola la “carpeta” donde se encuentran
los archivos .phy y .cfg
1. Presiona Enter para ejecutar el análisis..
50. Effective sample size
(ESS>200)
Convergencia de las distribuciones estacionarias
Tracer software
Tamaño efectivo de muestreo
de corridas no correlacionadas
independientes.
Tamaños menores a 200 en ESS,
significa alta correlación, por lo
que el muestreo no es
independiente (se marca en
rojo en Tracer).
52. Materiales y métodos |Práctica 1|
Revisión y edición de
secuencias (.ab1)
Alineamiento de
secuencias (.nex)
Modelo evolutivo de
sustitución de nucleótidos
Reconstrucción hipótesis
filogenética
Edición de árboles
http://www.geneious.com/
http://www.mbio.ncsu.edu/BioEdit/bioedit.html
http://phylemon.bioinfo.cipf.es/ev
olutionary.html
http://mrbayes.sourceforge.net/
http://jmodeltest.org/login http://www.robertlanfe
ar.com/partitionfinder/
https://www.phylo.org/portal2/login!input.action
https://code.google.com/arc
hive/p/garli/downloads
http://tree.bio.ed.ac.uk/software/figtree/
53. Árbol filogenético (.tre)
Abrir con Wordpad
A
B
C
D
E
A
B
C
D
E
A
B
C
D
E
Newick ((((A B) C) D) E) (((A B) (C D))E) ((A B) ((C D) E))
54. http://tree.bio.ed.ac.uk/software/figtree/
Edición de árboles
FigTree
1. File open
archivo.con.tre
2. Modificar los tamaños de
letra.
3. Asignar la especie para la raíz
a la rama de
ClongirostrisKU177803
4. Asignar los valores de
probabilidades posteriores
(pp) a los nodos.
5. Expanda o contraiga el árbol
de acuerdo a su interés.
55.
56.
57.
58. Ejercicio:
1. Utilice la barra de herramientas y el
panel de control para reproducir la
filogenia.
2. Exporte en formato .pdf y .jpg