
Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Similar a Lisosomas
Similar a Lisosomas (20)
Último
Último (20)
Lisosomas
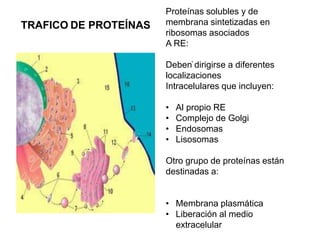
- 1. TRAFICO DE PROTEÍNAS . Proteínas solubles y de membrana sintetizadas en ribosomas asociados A RE: Deben dirigirse a diferentes localizaciones Intracelulares que incluyen: • Al propio RE • Complejo de Golgi • Endosomas • Lisosomas Otro grupo de proteínas están destinadas a: • Membrana plasmática • Liberación al medio extracelular
- 2. TRAFICO DE PROTEÍNAS . Cada proteína tiene una etiqueta que Permite su inclusión en la vesícula de transporte apropiada, la marca puede ser: 1. Una secuencia específica de aminoácidos 2. Una cadena lateral oligosacárido 3.Un dominio hidrófobo
- 3. TRAFICO DE PROTEÍNAS HACIA LOS ENDOSOMAS Y LISOSOMAS . Las enzimas lisosomales durante su viaje por RE y CG: 1. Sufren glicosilación 2. Eliminación de unidades de glucosa y manosa. 3.Fosforilación de las cadenas glucídicas lateralesoligosacáridos con manosa-6-P
- 4. LISOSOMAS vesícula membranosa de forma redondeada o polimorfa que contienen diferentes enzimas del tipo de hidrolasas ácidas (lipasas, nucleasas, proteasas, sulfatasas...) Estas enzimas hidrolíticas y proteolíticas sirven para digerir los materiales de origen externo (heterofagia) o interno (autofagia) que llegan a ellos. Es decir, se encargan de la digestión celular. Fueron descubiertos por De Duve en 1949 cuando realizaba estudios bioquímicos de la fosfatasa ácida en hepatocitos de rata.
- 5. El pH es de 5 en el interior del lisosoma. Para mantener este pH, los lisosomas poseen en su membrana una proteína transmembrana que bombea protones (H+) hacia el interior del lisosoma. El pH 5 es el óptimo para la actuación de las enzimas.
- 6. Función: Desde el punto de vista fisiológico se distinguen dos tipos básicos de lisosomas: 1) Lisosomas primarios: No participan en ningún proceso de digestión intracelular. Pueden verter sus enzimas al medio extracelular lisándolo, destruyendo células lesionadas o muertas (digestión extracelular).
- 7. Formación de lisosomas primarios Cada lisosoma primario es una vesícula que brota del aparato de Golgi, con un contenido de enzimas hidrolíticas (hidrolasas). Las hidrolasas son sintetizadas en el REG y viajan hasta el aparato de Golgi por transporte vesicular. Allí sufren una glicosilación terminal de la cual resultan con cadenas glucídicas ricas en manosa-6-fosfato (manosa 6-P). La manosa 6-P es el marcador molecular, la “estampilla” que dirige a las enzimas hacia la ruta de los lisosomas. Se ha estudiado una enfermedad en la cual las hidrolasas no llevan su marcador; las membranas del aparato de Golgi no las reconocen como tales y las empacan en vesículas de secreción para ser exocitadas.
- 8. 2) Lisosomas secundarios: Resultan de la fusión de un lisosoma primario con material de naturaleza variable, y están implicados en la digestión intracelular.
- 9. Según con el material con el que se fusionan se distinguen tres tipos de lisosomas secundarios: - Fagolisosomas ('vacuolas heterofágicas'): Resultan de la fusión con un fagosoma, el cual puede llevar partículas de gran tamaño, sustancias muy variadas e incluso bacterias o virus. Provocan la digestión intracelular del fagosoma y, de esta manera, la célula se defiende de agresiones patógenas o sustancias tóxicas. Se encuentran, por ejemplo, en los glóbulos blancos, capaces de fagocitar partículas extrañas que luego son digeridas en estos cuerpos.
- 10. Autofagolisosomas ('vacuolas autofágicas'): Resultan de la fusión con un autofagosoma, el cual procede de la envoltura del RE o de cualquier orgánulo o resto celular que ha de ser digerido (autodigerido). Intervienen en la digestión intracelular, obteniendo nutrientes necesarios para la vida de la célula. Participan también en los procesos de necrosis celular al digerir estructuras propias de la célula.
- 11. Endolisosomas: ('vacuolas heterofágicas'): Endosoma tardio Formados por la fusión con endosomas, es decir, vesículas procedentes de la endocitosis. Provocan su digestión intracelular para la obtención de nutrientes.
- 12. . Endosoma tardío: surge al unirse los lisosomas primarios con materiales provenientes de los endosomas tempranos. Los endosomas tempranos contienen macromoléculas que ingresan por los mecanismos de endocitosis inespecífica y endocitosis mediada por receptor. Este último es utilizado por las células para incorporar, por ejemplo, las lipoproteínas de baja densidad o LDL.
- 14. En todos los casos, en los lisosomas secundarios, a veces, permanece el material no degradado o digerido, después de la absorción originando los cuerpos residuales que, generalmente, son excretados. Un ejemplo de cuerpos residuales son los gránulos de lipofuscina que se observan en células de larga vida, como las neuronas.
- 15. La liberación de las hidrolasas cumple un papel fisiológico, permitiendo la reabsorción de estructuras que ya no son útiles, por ejemplo la cola de los renacuajos durante la metamorfosis.
- 16. Las enzimas lisosomales Las enzimas más importantes del lisosoma son: * Lipasas, que digiere lípidos * Glucosidasas, que digiere carbohidratos * Proteasas, que digiere proteínas * Nucleasas, que digiere ácidos nucleicos
- 18. Enfermedades lisosómicas Son enfermedades causadas por la disfunción de algún enzima lisosómico o por la liberación incontrolada de dichos enzimas en el citosol, lo que produce la lisis de la célula. En algunos casos, la liberación de las enzimas cumple un papel fisiológico, permitiendo la reabsorción de estructuras que ya no son útiles, por ejemplo la cola de los renacuajos durante la metamorfosis.
- 19. En las enfermedades de almacenamiento lisosómico, alguna enzima del lisosoma tiene actividad reducida o nula debido a un error genético y el substrato de dicho enzima se acumula y deposita dentro del lisosoma que aumentan de tamaño a causa del material sin digerir, lo cual interfiere con los procesos celulares normales; algunas de estas enfermedades son:
- 20. * Esfingolipidosis. Son enfermedades causada por la disfunción de alguno de los enzimas de la ruta de degradación de los esfingolípidos. Dado que los esfingolípidos abundan en el cerebro, varias de estas enfermedades cursan con retraso mental severo y muerte prematura; entre ellas hay que destacar: la enfermedad de Tay-Sachs, la enfermedad de Gaucher, la enfermedad de Niemann-Pick, la enfermedad de Krabbe.
- 21. * Carencia de lipasa ácida. La lipasa ácida es una enzima fundamental en el metabolismo de los triglicéridos y del colesterol, que se acumulan en los tejidos. La disfunción de esta enzima provoca dos enfermedades, la enfermedad de almacenamiento de ésteres de colesterol, en que la enzima presenta muy poca actividad y la enfermedad de Wolman, en que la enzima es totalmente inactiva.
- 22. * Glucogenosis tipo II o enfermedad de Pompe. Es un defecto de la α(1-4) glucosidasa ácida lisosómica, también denominada maltasa ácida. El glucógeno aparece almacenado en lisosomas. En niños destaca por producir insuficiencia cardíaca al acumularse en el músculo cardíaco causando cardiomegalia. En adultos el acúmulo es más acusado en músculo esquelético.
- 23. * Mucopolisacaridosis. Causadas por la ausencia o el mal funcionamiento de las enzimas necesarias para la degradación moléculas llamadas glicosoaminoglicanos o glucosaminglucanos (antes llamadas mucopolisacáridos). Destacan la mucopolisacaridosis tipo I, también conocida como gargolismo o enfermedad de Hurler, en la que existe un defecto de la enzima α-1-iduronidasa, y la mucopolisacaridosis de tipo II o síndrome de Hunter, causada por un error en la enzima iduronato-2-sulfatasa.
- 24. Gota En la gota, el ácido úrico proveniente del catabolismo de las purinas se produce en exceso, lo que provoca la deposición de cristales de urato en las articulaciones. Los cristales son fagocitados por las células y se acumulan en los lisosomas secundarios; estos cristales provocan la rotura de dichas vacuolas con la consiguiente liberación de enzimas lisosómicos en el citosol que causa la digestión de componentes celulares, la liberación de sustancias de la célula y la autolisis celular.
- 25. Artritis reumatoide La membrana de los lisosomas es impermeable a las enzimas y resistente a la acción de éstas. Ambos hechos protegen normalmente a la célula de una batería enzimática que podría degradarla. Existen, sin embargo, algunos procesos patológicos, como la artritis reumatoide, que causan la destrucción de las membranas lisosomales, con la consecuente liberación de las enzimas y la lisis celular.
- 26. ENFERMEDADES PRODUCIDAS POR DEFECTOS DE LA FUNCIÓN LISOSÓMICA. (Enfermedades por almacenamiento de Esfingolipidos)
- 27. ETAPAS DE DIGESTIÓN POR ENZIMAS DIGESTIVAS LISOSÓMICAS
- 28. VÍAS DEL SISTEMA LISOSOMICO
- 29. Secreción Las vesículas que brotan de la cara trans portan productos acabados destinados al medio extracelular. La fusión de dichas vesículas con la membrana plasmática –exocitosis- da como resultado la secreción o exportación de diversas sustancias: enzimas, hormonas, moléculas de la matriz extracelular o de la pared celular, anticuerpos y otras, según el tipo celular. Hay dos rutas secretorias: la continua o constitutiva y la discontinua o regulada.
- 30. La secreción continua o constitutiva está presente en todos los tipos celulares. Las vesículas que siguen esta ruta se exocitan en forma continua, a medida que brotan del aparato de Golgi. Por ejemplo, se secretan por esta vía las moléculas que se incorporan a la matriz extracelular.
- 31. La secreción regulada, en cambio, es propia de células secretoras especializadas. En estos casos, las vesículas se acumulan en el polo secretor de la célula, como gránulos de secreción, y la exocitosis se dispara sólo ante señales muy específicas. Por ejemplo, las células b de los islotes de Langerhans (en el páncreas), contiene gránulos de insulina que son exocitados en respuesta a una elevación de la glucemia.