
Recomendados
Más contenido relacionado
Similar a BIOLOGIA SEMANA 3 COMPLEMENMTACION.ppt
Similar a BIOLOGIA SEMANA 3 COMPLEMENMTACION.ppt (20)
Más de JEISSONDAVIDCABOSSAN
Más de JEISSONDAVIDCABOSSAN (11)
Último
Último (20)
BIOLOGIA SEMANA 3 COMPLEMENMTACION.ppt
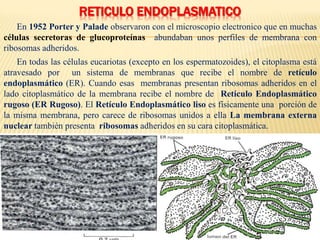
- 1. RETICULO ENDOPLASMATICO En 1952 Porter y Palade observaron con el microscopio electronico que en muchas células secretoras de glucoproteínas abundaban unos perfiles de membrana con ribosomas adheridos. En todas las células eucariotas (excepto en los espermatozoides), el citoplasma está atravesado por un sistema de membranas que recibe el nombre de retículo endoplasmático (ER). Cuando esas membranas presentan ribosomas adheridos en el lado citoplasmático de la membrana recibe el nombre de Retículo Endoplasmático rugoso (ER Rugoso). El Retículo Endoplasmático liso es físicamente una porción de la misma membrana, pero carece de ribosomas unidos a ella La membrana externa nuclear también presenta ribosomas adheridos en su cara citoplasmática.
- 2. Funciones del RER: En su interior se terminan de fabricar un gran numero de proteínas (glicoproteínas) de membrana y proteínas enzimáticas, así como otras que pasaran hacia las secciones del REL, donde serán utilizadas como “materias primas” para fabricar otros productos. El RER posee por su cara interna, en el interior de sus cisternas, múltiples enzimas responsables de esas actividades. Funciones del REL: Tiene en su interior, localizadas en su pared interna, enzimas responsables de: síntesis de fosfolípidos, lipoproteínas, colesterol y triglicéridos, así como otro grupo de ellas, para la destoxificación de productos provenientes del metabolismo oxidativo celular (radicales libres de oxigeno) y otras, encargadas de transformar muchas sustancias tales como medicamentos y tóxicos exógenos. También posee enzimas que liberan glucosa de los almacenes de glucógeno celular.
- 3. La membrana del REL incorpora constantemente fosfolípidos, colesterol, proteínas estructurales y enzimas que fabrica, a la estructura misma de sus membranas, por lo que crece constantemente formando protrusiones que se desprenden como vesículas de transporte del REL, que se dirigen flotando por el citosol hasta contactar con otro organelo membranoso: el aparato de Golgi.
- 4. APARATO DE GOLGI Es otro organelo membranoso situado generalmente muy cerca del núcleo y próximo al REL, pero separado de ambos. Sus membranas también tienen una constitución molecular semejante a la de la membrana celular. En la cara interna de sus cisternas existen numerosas enzimas encargadas de las diversas producciones de este organelo.
- 5. El Aparato de Golgi está formado por un grupo de cisternas aplanadas, sin ribosomas unidos a sus superficies, que presentan un sector, donde estas cisternas tienen una disposición convexa (región “cis”) y miran hacia las membranas del REL. El otro sector de cisternas tiene una disposición cóncava (región “trans”).
- 6. Por la región cis, Golgi recibe vesículas de transporte del REL, que se habían desprendido de este último conteniendo distintas lipo y glicoproteínas, las que ingresan a este sector del Golgi mediante fusión de sus membranas con las de la sección cis.
- 7. Golgi selecciona las distintas glico y lipoproteínas destinadas al citosol y a las membranas, les da el acabado final por glicosilación.
- 8. El sector trans del Golgi libera de sus cisternas cóncavas vacuolas, muchas de ella grandes, conteniendo los productos finales de este organelo. Algunas de estas vacuolas se fundirán con la membrana celular liberando su contenido al exterior, son los llamados gránulos de secreción. Otras vacuolas incorporan componentes a la membrana (glicoproteínas receptoras de membrana) para sus funciones. Otras transportan productos hacia el citosol.
- 10. LISOSOMAS : Estructura y Función PEROXISOMAS: Estructura y Función Biología Celular y Molecular Blgo. Ms.C. Jeisson Cabos Sánchez
- 11. LISOSOMA
- 12. CARACTERÍSTICAS son orgánulos relativamente grandes, formados por el retículo endoplasmático rugoso y luego empaquetadas por el complejo de Golgi, que contienen enzimas hidrolíticas y proteolíticas que sirven para digerir los materiales de origen externo (heterofagia) o interno (autofagia) que llegan a ellos. Es decir, se encargan de la digestión celular. La degradación de micromoléculas en sus componentes y la transferencia de éstos al citoplasma, permite a la célula su reutilización para síntesis de nuevas macromoléculas se les define como vesículas formadas por membrana simple , tamaño es variable (0,2 a 2mm).
- 13. Lisosomas • Organela ligada a la membrana, que es responsable por la degradación de las proteínas y membranas en la célula. • También ayuda a degradar materiales ingeridos por la célula. • Función: digestión celular
- 14. Las enzimas del lisosoma • Las hidrolasas ácidas son enzimas hidrolíticas que están activas en condiciones ácidas. • El lumen se mantiene a un pH ácido mediante la acción de una bomba de H+ situada en la membrana.
- 15. Hidrolasas Acidas a) Glicosidasas: enzimas lisosomales involucradas en degradación de cadenas glicosídicas presentes en glicoproteínas, esfingolípidos, glucógeno, o mucopolisacáridos. Glucosidasa alfa y beta , Manosidasa, Galactosidasas, Hialuronidasa, Lisozima, Sulfatasas (aril sulfatasa). b) Proteasas: enzimas lisosomales involucradas en la hidrólisis de cadenas peptídicas: catepsinas, carboxipeptidasas, aminopeptidasas. c) Lipasas: enzimas que hidrolizan esfingolípidos, esteres de ácidos grasos y fosfoglicéridos: esfingomielinasa, ceraminidasa, lipasa etc.
- 16. d. Fosfatasas: enzimas que hidrolizan grupos fosfato presentes en ácidos nucleicos , proteínas, lípidos y carbohidratos: fosfatasa ácida, fosfolipasas, exonucleasa ácida, nucleotidasa ácida, esfingo fosfodiesterasa e. Nucleotidasas: hidrolizan ácidos nucleicos: endonucleasas, desoxirribonucleasa, ribonucleasa.
- 17. Tipos de lisosomas: • Los lisosomas primarios son aquellos que sólo contienen las enzimas digestivas, mientras que los lisosomas secundarios, por haberse fundido con una vesícula con materia orgánica, contienen también sustratos en vía de digestión: vacuolas digestivas o heterofágicas, cuando el sustrato procede del exterior, y vacuolas autofágicas, cuando procede del interior
- 19. AUTOFAGIA
- 20. FORMA DE OBTENCIÓN DE MATERIALES DE DEGRADACIÓN 1. ENDOCITOSIS. Toma de sustancia por formación de vesícula de membrana plasmática 2. FAGOSOMA: fagocitosis , vesículas que carecen de enzimas hidrolíticas 3. AUTOGAGIA: digestión de material celular por sus propias enzimas.
- 21. FUNCIONES DE LOS LISOSOMAS 1.Digestión celular ( Carbohidratos ---- > monosacaridos; proteínas ---- > dipeptido ---- > aminoácidos que quedan en el citosol) 2. Digestión de material extracelular ( microorganismos, macromoléculas exógenas, hidrolasas son excretadas por exocitosis) 3. Autofagia: cumplimiento de ciclo celular ó recambio de componentes celulares: organelas ( reabsorción y formación de tejido óseo, proceso de fecundación del ovulo, durante determinadas etapas de la morfogénesis en las que hay invasión celular
- 22. Importancia de los lisosomas • Defensa: papel ejecutado por leucocitos y macrófagos. • Procesos patológicos: artritis reumatoide (falta fosfotransferasa, la secreción de hidrolasas produce daño a los cartilagos) – La proteasa del acrosoma del espermatozoide destruye el trayecto que conduce a la superficie del huevo – Acumulación de ácido úrico, produciendo inflamación en los tejidos
- 23. Sindrome de Hunter • La mucopolisacaridosis tipo II o síndrome de Hunter, causada por un error en la enzima iduronato-2-sulfatasa. Produce también alteraciones en el fenotipo y en el desarrollo físico y mental del niño
- 24. Enfermedad de Gaucher • que se debe a la deficiencia de la enzima betaglucosidasa ácida, que origina un depósito de un glucocerebrósido, la glucosilceramida, en el sistema retículoendotelial.
- 25. Enfermedad de TAY SACHS (GM2) • Causada por una deficiencia de una enzima llamada HEXOSAMINIDASA, esta enzima tiene la función de degradar sustancias grasas llamadas gangliósidos. • A consecuencia de esto a la célula tiene sobrepoblación de estos gangliòsidos y no puede llevar a cabo sus funciones normales. • Esto es muy peligroso ya que los gangliósidos son componentes fundamentales del plasmalema neuronal. • Sintomas: Apatía, ceguera, ligero retraso mental, irritabilidad convulsiones, disminución del tono muscular, sordera, demencia , parálisis , degeneración progresiva del sistema nervioso central
- 26. Enfermedad de Sanfilipo • El síndrome de Sanfilippo, o Mucopolisacaridosis (MPS) tipo lll , comprende un grupo de enfermedades de almacenamiento lisosomal. • Causada por la deficiencia de una de las cuatro hidrolasas lisosomales que participan en la degradación del glicosaminoglicano Heparan Sulfato (el cual se encuentra localizado en la matriz extra-celular y en las glicoproteínas de la superficie celular). • Esta deficiencia ocasiona degeneración severa del sistema nervioso central y deterioro de las habilidades sociales y de adaptación
- 27. Enfermedad de Pompe • Glucogenosis tipo II. • Es un defecto de la α(1-4) glucosidasa ácida lisosómica,(GAA) también denominada maltasa ácida. • El glucógeno aparece almacenado en lisosomas. • Enfermedad muscular debilitante y rara que afecta a niños y adultos. • Forma infantil y juvenil. • Se caracterizan generalmente por un debilitamiento muscular progresivo y dificultades respiratorias, pero la gravedad de la enfermedad puede variar ampliamente dependiendo de la edad de inicio y cuán afectados se encuentren los órganos
- 28. Otras enfermedades lisosomales Artritis reumatoide Gota
- 29. PEROXISOMAS
- 30. Peroxisomas • Los peroxisomas se denominan así por que generalmente contienen una o mas enzimas que utilizan oxígeno molecular para eliminar átomos de hidrógeno a partir de susbstratos orgánicos específicos (R) a través de una reacción oxidativa que produce peróxido de hidrógeno (H2O2) RH2 + O2 ---- R + H2O2
- 32. Peroxisomas = Microcuerpos • Organelos autoreplicables. Presentan enzimas oxidativas (catalasa, uratoxidasa). • Presenta en hígado y riñón como gránulos (0,6 μm) que posee membrana simple y matriz densa. Crece con lentitud y es destruido por autofagia. • Lugares de oxidación del oxígeno por reacciones oxidativas: producen peróxidos de hidrogeno. • Presente en la mayoría de células eucariotas de mamíferos. Importancia • Formar y descomponer peroxido de Hidrogeno. Participan en la beta oxidación de ácidos grasos. Detoxificar a la célula (etanol). Lugares de degradación de las purinas. Metabolismo de trigliceridos • Envejecimiento de la célula (radicales libres afectan ADN, membrana) • Permite la acción de la superoxido dismutasa • En las plantas : fotorrespiración
- 33. Síndrome Zellweger • síndrome cerebro- hepato-renal. • Desorden congénito . • Baja producción o ausencia de producción de peroxisomas. • Descrito por Hans Ulrich Zellweger
- 34. MITOCONDRIA: ESTRCTURA Y FUNCIÓN Se describen generalmente como generadores de energía de las células ya que producen la mayor parte del ATP necesario. Su tamaño varía entre 0.5-10 micrómetros. Su estructura esta basada en: Membrana externa Membrana interna Espacio intermembranoso Matriz mitocondrial
- 35. MEMBRANA INTERNA Contiene mas proteínas que la membrana externa, se considera altamente selectiva ya que solo permite el paso de metabolitos como el ATP, ADP, piruvato, Ca+ y fosfato.
- 36. Esta membrana esta dividida en pliegues que forman las crestas mitocondriales que se proyectan en dirección hacía la matriz y aumentan en gran medida la superficie, donde puede llevarse a cabo la síntesis del ATP. Las mitocondrias contienen ADN y ARN propios, además de ribosomas para la biosíntesis de proteínas en su matriz. Pero la mayoría de las proteínas que usa son nucleares, ya que solo produce un 5% del total que necesita.
- 37. En ella se produce la cadena de transporte de electrones, compuesta por cuatro complejos enzimáticos fijos y dos transportadores de electrones móviles: 1.Complejo I o NADH deshidrogenasa. 2.Complejo II o succinato deshidrogenasa; ambos ceden electrones al coenzima Q o ubiquinona. 3.Complejo III o citocromo b-c1 que cede electrones al citocromo c. 4.Complejo IV o citocromo c oxidasa que cede electrones al O2 para producir dos moléculas de agua.
- 38. También tiene un complejo enzimático que cataliza la síntesis de ATP, al que se le conoce como fosforilación oxidativa. Se conforma de proteínas transportadoras que permiten el paso de iones y moléculas.
- 40. • Define el perímetro exterior liso de las mitocondrias. • Contiene porinas. Proteinas transportadoras transmembrana que forman canales acuosos a través de la bicapa lipídica.
- 41. ESPACIO INTERMEMBRANOSO Se encuentra entre la membrana interna y externa, en el se encuentra una alta concentración de protones como resultado del bombeo de los mismos. En el se localizan diversas enzimas que intervienen en la transferencia del ATP.
- 42. MATRIZ MITOCONDRIAL Se encuentra dentro de la membrana interior Aproximadamente 50% agua En ella tienen lugar diversas rutas metabólicas como el ciclo de Krebs y la beta-oxidacion de ácidos grasos
- 43. FOSFORILACION OXIDATIVA • Es la transferencia de electrones de los equivalentes reducidos NADH y FADH, obtenidos en la glucólisis y en el ciclo de Krebs incluyendo el oxígeno molecular, todo esto acoplado con la síntesis de ATP. • Este proceso metabólico está formado por un conjunto de enzimas complejas, ubicadas en la membrana interna de las mitocondrias, que catalizan varias reacciones de óxido- reducción, donde el oxígeno es el que acepta al final los electrones y donde se forma finalmente agua.
- 45. Es el principal organelo celular, contiene el material genético constituido por ADN junto con proteínas especiales llamadas histonas. El núcleo es generalmente grande, posee una membrana porosa y en su interior se encuentra el ADN como una maraña de hilos delgados, llamada cromatina. Cuando la célula comienza su proceso de división (cariocinesis), la cromatina se condensa y los cromosomas se hacen visibles como entidades independientes. El cromosoma es el material hereditario cuya principal función es conservar, transmitir y expresar la información genética que contiene. Rodeado de carioteca formado por bicapa de fosfolípidos. Presenta poros nucleares (proteínas). Presenta nucléolos en donde se fabrican las subunidades de los ribosomas. Lleva la información hereditaria y ejerce influencia sobre las actividades celulares.
- 46. NUCLEO INTERFÀSICO Definición: El núcleo interfásico es la estructura que tiene el núcleo durante la interfase, es decir cuando la célula no se divide, en esta etapa su actividad metabólica es máxima.
- 47. ESTRUCTURA DEL NÚCLEO CELULAR Envoltura Nuclear Es una doble bicapa lipídica. Presenta Poros nucleares, permite el intercambio de moléculas entre el citoplasma y el nucleoplasma Nucleoplasma Está formado por: Agua, enzimas, electrolitos, etc., en él se encuentra el ADN (cromatina) Cromatina Estructura formada por ADN, en ellos se encuentran los genes (Unidades genéticas hereditarias). Nucleolo Estructura nuclear formada por ADN y proteínas, donde se sintetizan los distintos tipos de ARN.
- 48. ESTRUCTURA
- 49. La envoltura nuclear (carioteca) es una doble membrana (externa e interna) y entre ambas membranas queda un espacio de 25-40 nm, que constituye la llamada cisterna peri-nuclear que se comunica con la cavidad del retículo endoplasmático. Presenta poros nucleares, en los que ambas membranas se fusionan y quedan interrumpidas, de trecho en trecho, estableciéndose comunicaciones entre el citoplasma y el nucleoplasma. ENVOLTURA NUCLEAR
- 50. Los poros son abundantes, en células embrionarias, en células inmaduras y, en general en células muy activas, que necesitan un grado considerable de transferencia entre núcleo y citoplasma. La estructura del poro comprende al denominado complejo del poro, que consta de: a. Ocho columnas proteicas b. Proteínas de anclaje que amarran las columnas proteicas a la envoltura nuclear. c. Proteínas radiales que surgen de las columnas y se orientan hacia el centro. d. Fibrillas proteicas que nacen de las bocas internas y externas del complejo del poro. PORO NUCLEAR
- 51. LAMINA NUCLEAR • Estructura protéica formada por filamentos intermedios del citoesqueleto, participa en la división celular y dá forma al núcleo celular. • La lámina nuclear es una delgada malla de laminofilamentos (A, B y C) entrecruzados. • Del lado interno la lámina B se une a la envoltura nuclear y las láminas A y C se fijan a puntos específicos de la cromatina y guían las interacciones de la cromatina. • Le otorga resistencia a la carioteca y establece su forma generalmente esférica. • La envoltura nuclear y la lámina nuclear se desarman al comienzo de la mitosis.
- 52. LA CROMATINA La cromatina es el componente más abundante del núcleo y está constituido por DNA unido a proteínas del tipo histonas. Son 5 histonas: la H1 y las 4 histonas nucleosómicas, que son H2A, H2B, H3 y H4. Cromatosoma
- 53. • En un nucleosoma, las histonas nucleosómicas se asocian y forman una estructura octamérica, el núcleo del nucleosoma, alrededor de ellas se enrolla el DNA. • El complejo formado por el nucleosoma más la histona H1 se le denomina cromatosoma. • Los cromatosomas se enrollan sobre sí mismos y dan lugar a una estructura helicodidal llamada solenoide, de 30 nm. de diámetro. • El enrollamiento depende de las histonas H1, y cada vuelta del solenoide y sólo contiene seis nucleosomas.
- 54. • La fibra de 30 nm se organiza en una serie de asas muy amplias superenrolladas o dominios, que pueden compactarse en fibras aún más gruesas (80 a 100nm) • Las asas de DNA se unen en sus extremos con proteínas que forman parte de un andamiaje nuclear organizado. • Los cromosomas mitóticos representan la última etapa del empaquetamiento de la cromatina.
- 55. EUCROMATINA Y HETEROCROMATINA • La eucromatina corresponde a la cromatina desespiralizada y, por tanto transcripcionalmente activa, mientras que la heterocromatina sería la cromatina muy espiralizada o condensada y, en principio transcripcionalmente inactiva, pues la condensación hace que el DNA no sea accesible a las proteínas activadoras de los genes. Se distinguen 2 tipos de heterocromatina: - Constitutiva, cromatina altamente condensada que se encuentra de manera constante en todos los tipos celulares - Facultativa, la que se detecta en localizaciones que varían en los distintos tipos celulares.
- 56. Centro fibrilar: complejo de nucleoproteínas regulatorias que dirigen la transcripción tal como RNA Pol I y DNA topoisomerasa I. Componente fibrilar denso: región de alta densidad electrónica. Contiene DNA, RNAr naciente y proteínas asociadas. Región granular: precursores ribosomales maduros (RNAr 28s y 18s) e interme- diarios en ensamble de los ribosomas. NUCLEOLO Son estructuras esféricas y densas que se tiñen intensamente, con un diámetro de 1-3 m. Frecuentemente el nucleólo es único, aunque existen núcleos con dos o más nucleolos.
- 57. FUNCIONES DEL NÚCLEO - Controlar la expresión genética . - Mediar en la replicación del ADN durante el ciclo celular. - En él se realiza la transcripción y el procesamiento del ARN mensajero
- 58. ACIDOS NUCLEICOS • Los ác. nucleicos son las moléculas que contienen la información que prescribe la secuencia de aminoácidos en las proteínas • Todos los organismos vivos contienen ácidos nucleicos en forma de ácido dexosirribonucleico (DNA) y ácido ribonucleico (RNA). • Algunos virus sólo contienen DNA, mientras que otros sólo poseen RNA. • EL DNA y el RNA tienen grandes semejanzas químicas. Por sus estructuras primarias ambos son polímeros lineales compuestos por monómeros denominados nucleótidos
- 59. ACIDOS NUCLEICOS, son polímeros de nucleótidos Enlace N-glucosídico Enlace fosfoéster Un nucleótido está formado por 3 elementos: una base nitrogenada, una pentosa y un grupo fosfato.
- 60. Las bases nitrogenadas de los desoxirribonucleótidos del ADN son:
- 61. Apareamiento de bases nitrogenadas en el ADN es: Adenina con Timina (2 puentes de H) y Citosina con Guanina (3puentes de H) 2 Puentes de H 3 Puentes de H
- 62. Los nucleótidos se unen para formar ácidos nucléicos, mediante enlaces fosfodiéster
- 63. Modelo de doble hélice (James Watson y Francis Crick –1953) Dos fuentes de información: 1. Estudio de composición de bases de Erwin Chargaff • El DNA es una doble cadena que consiste de ~50% purinas (A,G) y ~50% pirimidinas (C, T) • La cantidad de A=T y la cantidad de G=C (regla de Chargaff) • % GC varía de organismo a organismo. Ejemplos: %A %T %G %C %GC Homo sapiens 31.0 31.5 19.1 18.4 37.5 Zea mays 25.6 25.3 24.5 24.6 49.1 Drosophila 27.3 27.6 22.5 22.5 45.0 Mycobacterium sp. 12.0 11.0 28.0 26.0 ACIDO DESOXIRRIBONULEICO
- 64. ESTRUCTURA SECUNDARIA DEL ADN • Es una doble hélice de 2 nm de diámetro. 2 nm Par de bases nitrogenadas • Las bases nitrogenadas se encuentran en el interior. • Las parejas de bases se encuentran unidas a un armazón formado por las pentosas y los grupos fosfato. Armazón fosfoglucídico • El enrollamiento es dextrógiro y plectonémico. • Cada pareja de nucleótidos está situada a 0,34 nm de la siguiente y cada vuelta de doble hélice contiene 10 pares de nucleótidos. 3,4 nm 0,34 nm • Las dos cadenas son antiparalelas y complementarias. WATSON Y CRICK ,1953 CHARGAFF: Contenido púricas= pirimidínicas
- 67. ACIDO RIBONUCLEICO • Es una simple cadena • Tiene ribosa • Tiene Uracilo en lugar de Timina • No se cumple la regla de Chargaff. Estructura secundaria: Estructura terciaria:
- 68. TIPOS DE ARN ARN mensajero Determina la secuencia de aminoácidos en la TRADUCCIÓN ARN ribosomal Participa en la Traducción o síntesis proteica ARN de transferencia Activa los aa y los incorpora a la nueva proteína durante la traducción. ARNsn pequeño nuclear Participa en el procesamiento del transcripto primario
- 69. ARN MENSAJERO ADN ARN mensajero Su función es copiar la información genética del ADN y llevarla hasta los ribosomas. En eucariotas porta información para que se sintetice una proteína: MONOCISTRÓNICO. En procariotas contiene información separada para la síntesis de varias proteínas distintas: POLICISTRÓNICO. Tiene una vida muy corta (algunos minutos) ya que es destruido rápidamente por las ribonucleasas.
- 70. ARN DE TRANSFERENCIA 3’ 5’ Brazo T Brazo A Brazo D Anticodón Transportan los aminoácidos hasta los ribosomas. Todos los tipos de ARNt comparten algunas características: En el extremo 5’ un triplete que tiene guanina y un ácido fosfórico libre. En el extremo 3’ tres bases (C-C-A) sin aparear. Por este extremo se une al aminoácido. En el brazo A un triplete de bases llamado anticodón diferente para cada ARNt en función del aminoácido que transportan. Zona de unión a la enzima que lo une al aminoácido. Zona de unión al ribosoma. Zona de unión al ARNm.
- 71. RESUMEN
- 74. CROMOSOMAS • El ADN (acido desoxirribonucleico) es una biomolécula orgánica (un ácido nucleico), formado por nucleótidos de adenina, timina, citosina y guanina. • Etimológicamente Cromosoma proviene del termino griego (chroma= color y soma= cuerpo o elemento).
- 78. • La unidad básica de información genética es el gen • GEN: elementos que contienen la información que determina las características de una especie.
- 79. CARIOTIPO Conjunto de cromosomas de una célula agrupados por pares homólogos y dispuestos según un orden preestablecido. Para ordenarlos se tiene en cuenta la longitud, forma, posición del centrómero y las bandas observables. En la especie humana, la observación del cariotipo permite detectar anomalías genéticas importantes, como el síndrome de Down (trisomía en el cromosoma 21).
- 80. cariotipo
- 82. Defectos cromosómicos más frecuentes • Síndrome de Down: Se denomina así a la trisomía 21, un defecto genético en que el cromosoma 21 aparece triplicado; sus manifestaciones se asocian al retardo metal y cambios físicos. • Síndrome de Edwars: Es la trisomía 18 en que aparece este cromosoma triplicado; sus manifestaciones son más graves que las de la Trisomía 21. • Síndrome de Patau: Aparece un cromosoma extra en el cromosoma 13; conlleva defectos y retardo mental aún más severos