Descargar para leer sin conexión









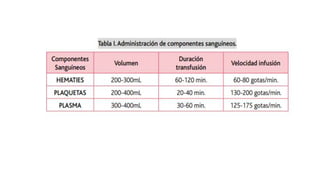
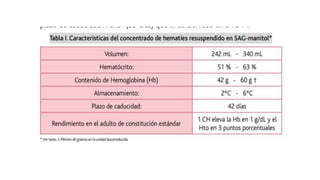


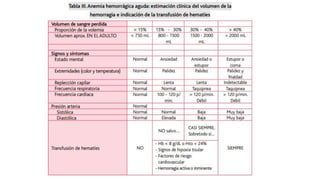


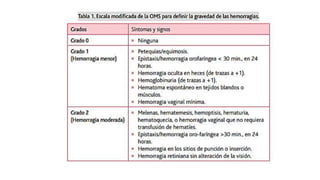





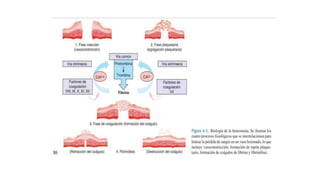

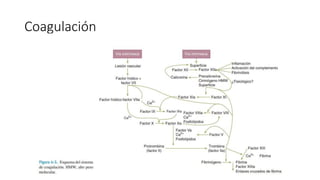












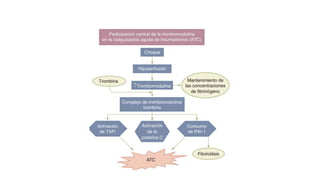
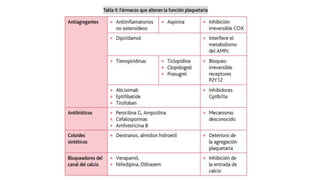

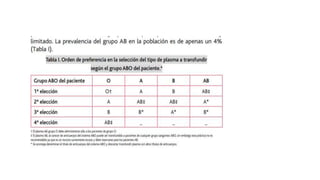












El documento habla sobre la medicina transfusional, que comprende procesos para obtener componentes sanguíneos. Describe brevemente la historia de la transfusión sanguínea y el desarrollo de conceptos como los grupos sanguíneos. Explica los tipos de concentrados eritrocitarios, plaquetarios y plasma, así como indicaciones y criterios para la transfusión. También aborda temas como la hemostasia, deficiencias de factores de coagulación, y complicaciones asociadas a la transfusión y la anticoagulación.





























![TEP [Autoguardado].pptxKLFRKLFKLFKLFKLKFLF](https://cdn.slidesharecdn.com/ss_thumbnails/tepautoguardado-240617182244-2ad9d0bf-thumbnail.jpg?width=640&height=640&fit=bounds)







