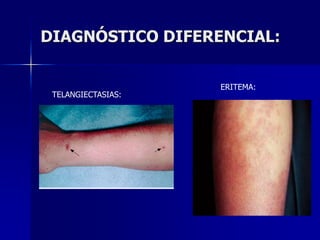
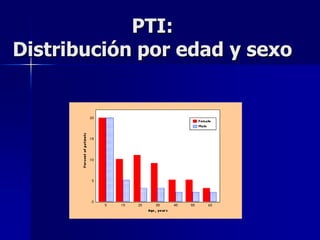
Este documento describe los síndromes púrpuricos, que se caracterizan por sangrados cutáneos y mucosos debidos a trastornos de la hemostasia primaria. Explica las púrpuras trombocitopénicas y no trombocitopenicas, haciendo énfasis en la púrpura trombocitopenia inmune, su patogenia, diagnóstico y tratamiento. Resume los diferentes tipos de tratamiento disponibles para esta enfermedad, incluyendo corticoides, inmunoglobulina y es