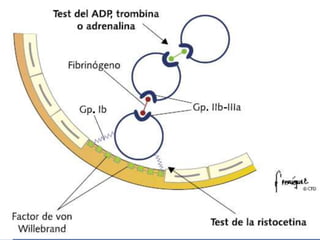
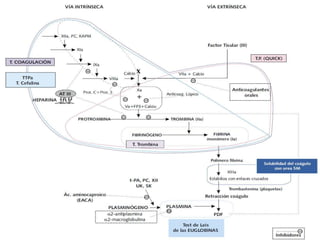
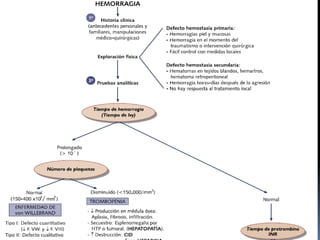
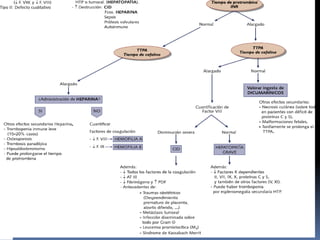
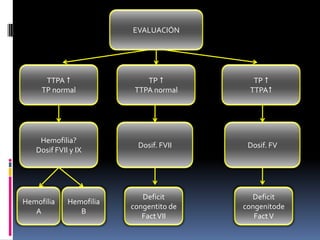
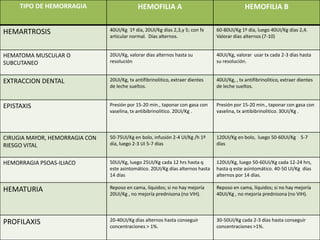
Este documento describe los trastornos de la hemostasia en pediatría. Resume los conceptos básicos de la hemostasia, los principales métodos diagnósticos de los trastornos hemorragíparos y las pautas de aproximación ante un paciente con sospecha de trastorno de la coagulación. Describe los trastornos hemorragíparos más frecuentes en edad pediátrica como la PTI, las hemofilias y la Enfermedad de Von Willebrand.