
Recomendados
Recomendados
Más contenido relacionado
Similar a Radiología de los desórdenes metabólicos del SNC.pptx
Similar a Radiología de los desórdenes metabólicos del SNC.pptx (20)
Último
Último (9)
Radiología de los desórdenes metabólicos del SNC.pptx
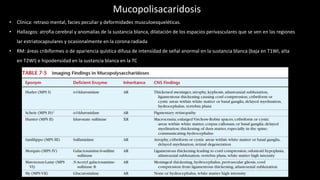
- 1. Mucopolisacaridosis • Clínica: retraso mental, facies peculiar y deformidades musculoesqueléticas. • Hallazgos: atrofia cerebral y anomalías de la sustancia blanca, dilatación de los espacios perivasculares que se ven en las regiones lar estriatocapsulares y ocasionalmente en la corona radiada • RM: áreas cribiformes o de apariencia quística difusa de intensidad de señal anormal en la sustancia blanca (baja en T1WI, alta en T2WI) e hipodensidad en la sustancia blanca en la TC
- 3. Lipidosis y otras enfermedades de depósito Las gangliosidosis son enfermedades que manifiestan anomalías de la sustancia blanca y atrofia cortical
- 5. Defectos mitocondriales Las gangliosidosis son enfermedades que manifiestan anomalías de la sustancia blanca y atrofia cortical Cuando las mitocondrias son defectuosas, las células no tienen suficiente energía. Las moléculas de oxígeno y combustible no utilizadas se acumulan en las células, causando daños.
- 6. Defectos mitocondriales Enfermedad de Leigh (Encefalomielopatía necrotizante subaguda) • Deficiencia en las enzimas asociadas con el desglose de piruvato. • Clínica: ataxia, nistagmo, oftalmoplejía, espasticidad, retraso psicomotor, parálisis craneales y acidosis metabólica. • Observado en niños menores de 2 años y expresada en RM por intensidad de señal alta del putamen bilateral en T2WI • El hallazgo de laboratorio revelador es la acidosis metabólica con niveles elevados de lactato.
- 7. Defectos mitocondriales Síndrome de MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, and Strokelike Episodes Syndrome) Hallazgos: • Múltiples áreas de anormalidad cortical de alta intensidad de señal en T2WI, que a menudo cruza la distribución de una arteria cerebral tradicional. • Atrofia con predominio en parte posterior del cerebro. • También puede afectar al putamen, caudado y tálamos. • Las lesiones pueden desaparecer con el tiempo, dejando sólo una mínima dilatación del surco • Aunque las lesiones pueden ser brillantes en DWI, por lo general NO muestran valores de coeficiente de difusión aparente (ADC) reducidos y, por lo tanto, no son infartos agudos Los pacientes pueden tener ceguera cortical Niveles elevados de ácido láctico
- 9. Defectos mitocondriales Síndrome de Kearns-Sayre Afectación de: • Órbitas: retinitis pigmentosa, oftalmoplejía, debilidad de los músculos extraoculares (y posible atrofia) • Corazón: déficits de conducción cardiaca y miocardiopatía. Hallazgos frecuentes: • Ganglios basales hiperintensos • Atrofia cerebral y cerebelosa • Hiperintensidad difusa de la sustancia blanca
- 10. Defectos mitocondriales Síndrome de Zellweger Trastorno autosómico recesivo con el gen defectuoso en el cromosoma 8 Caracterizado por: • Hipomielinización • Malformaciones corticales microgíricas y paquigíricas que son más graves en el perisilvio y regiones perirrolándicas, así como el surco caudotalámico • Quistes • Calcificaciones de tejidos blandos • Ensanchamiento sutural y macrocefalia.
- 11. Aminoacidopatías y otras deficiencias enzimáticas Mas frecuente en niños • Se manifiesta con retraso en el desarrollo, retraso mental, convulsiones, y vómitos. • La maduración normal del cerebro a menudo se retrasa, y la mielinización puede verse afectada. • La atrofia cerebral se ve en las últimas etapas
- 12. Enfermedad de Krabbe (Leucodistrofia de células globoides) • Deficiencia de galactocerebrósido-beta galactocerebrosidasa. • Gen en el cromosoma 14. • Acumulación dañina de galactosilesfingosina cerebrosido y psicosina en el parénquima cerebral. • Forma infantil (menor a 2 años), afectación de: • Tractos piramidales • Sustancia blanca cerebelosa • Sustancia gris profunda • Cuerpo calloso posterior • Sustancia blanca parietooccipital. • Forma de inicio tardío, similares lugares de afectación a excepción de sustancia blanca del cerebelo y la sustancia gris profunda. • Colina y mioinositol anormalmente elevados • La atrofia finalmente se desarrolla
- 13. Acidemia glutárica Hallazgos: • Macrocefalia con opérculos abiertos • Alta señal anormal en los ganglios basales • Datos de encefalitis. • Atrofia cerebral • Dilatación del espacio subaracnoideo anterior a los lóbulos temporales • Pérdida de volumen de los ganglios basales
- 14. Enfermedad de la orina de jarabe de mapple Secundaria a anomalías descarboxilación de aminoácidos de cadena ramificada Hallazgos: • US transfontanelar: aumento simétrico de la ecogenicidad de sustancia blanca periventricular, ganglios basales, principalmente globus pálido y tálamos. • La TC y la RM muestran: edema difuso en lugares similares, así como en el cerebelo y la cápsula.
- 15. Homocistinuria Deficiencia de cistationina beta sintasa, mapeada en el cromosoma 21. Múltiples eventos trombóticos, incluidos los accidentes cerebrovasculares. La presencia de subluxaciones del cristalino podría sugerir un diagnóstico de síndrome de Marfan, pero aquellos con homocistinuria están "arriba y adentro" y aquellos con enfermedad de Marfan están "abajo y afuera". Hallazgos: atrofia óptica, osteoporosis, cataratas, escoliosis, y cuerpos vertebrales bicóncavos.
- 16. Acidemia propiónica (PPA) y acidemia metilmalónica (MMA) Trastorno hereditario del ciclo tricarboxílico que genera acidosis. Síntomas: dificultades para alimentarse, hipotonía muscular, coreoatetosis, microcefalia y convulsiones. La enfermedad ocurre con más frecuencia en pacientes de Arabia Saudita. Hallazgos: • Atrofia y señal anormal de sustancia blanca, por retrasos en la mielinización (RMN) • Afectación de ganglios basales (típicamente en el globo pálido en MMA y en el globo pálido, caudados y putamina en PPA)
- 17. Desórdenes lisosomales Lipofuscinosis ceroide neuronal juvenil • Acumulación de lipopigmento en neuronas. • Pérdida de volumen del SNC, más prominente en el cerebelo • Tálamo hipointenso y sustancia blanca periventricular hiperintensa en T2WI
- 18. Capítulo 8 ANOMALÍAS CONGÉNITAS DEL SISTEMA NERVIOSO CENTRAL
- 19. Momentos de formación de lesiones congénitas El sistema nervioso central pasa por seis eventos principales de desarrollo que incluyen: 1. Neurulación primaria (3 a 4 semanas de gestación) 2. Desarrollo promesencefálico (2 a 3 meses de gestación) 3. Proliferación neuronal (3 hasta los 4 meses de gestación) 4. Migración neuronal (3 a 5 meses de gestación) 5. Organización (5 meses de gestación a años postnatales) 6. Mielinización (desde el nacimiento hasta los años posteriores al nacimiento).
- 20. Neurulación primaria y secundaria Neurulación primaria tienen lugar en la cara dorsal del embrión y da como resultado el desarrollo del cerebro y la médula espinal. • La primera fusión de los pliegues neurales se produce en la parte inferior del bulbo raquídeo. El cierre generalmente procede rostral y caudalmente. • Trastornos de primaria neurulación son: craneosquisis, anencefalia, mielosquisis, encefalocele, mielomeningocele y malformaciones de Chiari II. La neurulación secundaria ocurre más tarde, con procesos secuenciales de canalización y diferenciación regresiva. • Los trastornos de la neurulación secundaria son: malformaciones del sacro inferior y segmentos coxígeos del tubo neural que afectan al cono medular, filum terminale y cauda equina.
- 21. Desarrollo promesencefálico La relación inductiva principal se da entre el mesodermo precordal y el mesodermo de la notocorda y el prosencéfalo. Durante este evento se produce la formación de la cara y el cerebro anterior • Anomalías faciales (como ciclopía y probóscide) • Malformaciónes del cerebro (Aprosencefalia, atelencefalia, holoprosencefalia , agenesia del cuerpo calloso, agenesia del septum pellucidum, displasia septoóptica, displasia hipotalámica)
- 22. Proliferación neuronal Todas las neuronas y células gliales se derivan de las zonas ventricular y subventricular de la matriz germinal. Los trastornos de la proliferación neuronal pueden dar lugar a un cerebro pequeño o grande (microcefalia o macrocefalia).
- 23. Migración neuronal Las neuronas migran desde las zonas ventricular y subventricular del subepéndimo a su residencia final para la vida. Dos tipos de migración celular: radial y tangencial. • La migración radial conduce a las neuronas de la corteza cerebral y núcleos profundos en el cerebro y células de Purkinje en el cerebelo. • La migración tangencial conduce a las interneuronas de la corteza cerebral y capa granular interna de la corteza cerebelosa. Los trastornos de la migración neuronal incluyen esquizencefalia, lisencefalia (paquigiria), polimicrogiria, heterotopía y disgenesia cerebrocortical focal.
- 24. GRACIAS
Notas del editor
- Clínicamente, las mucopolisacaridosis (MPS) se caracterizan por retraso mental, facies peculiar y deformidades musculoesqueléticas. Las mucopolisacaridosis se asocian con atrofia cerebral y anomalías de la sustancia blanca en la imagen (tabla 7-5). Los diagnósticos de estos trastornos generalmente se basan en la evaluación bioquímica y/o cromosómica. Las mucopolisacaridosis suelen ser se manifiesta en la RM como áreas cribiformes o de apariencia quística difusa de intensidad de señal anormal en la sustancia blanca (baja en T1WI, alta en T2WI) e hipodensidad en la sustancia blanca en la TC (fig. 7-37). A menudo hay dilatación de los espacios perivasculares que se ven en las regiones lar estriatocapsulares y ocasionalmente en la corona irradian blanco materia. En el foramen magnum, engrosamiento de la duramadre (o depósito de mucopolisacáridos) puede causar compresión medular en algunos de los mucopolisacáridos. Algunos de estos trastornos están asociados con agrandamiento cabezas y cráneos engrosados. MPS III, enfermedad de Sanfilippo, es único entre las mucopolisacaridosis en que el las anomalías no relacionadas con el SNC son relativamente menores. Aracnoides Los quistes se incluyen entre los hallazgos intracraneales con Trastornos MPS. Se supone que la acumulación de glicosaminoglicanos en las leptomeninges explica la obstrucción de la membrana aracnoidea y el efecto de válvula de bola que puede conducir a tales quistes, así como a la dilatación de la vaina del nervio óptico.
- FIGURA 7-37 Mucopolisacaridosis de Hunter. A, la exploración FLAIR muestra espacios perivasculares dilatados (hipointenso) y sustancia blanca anormal de la corona radiada (hiperintenso). B, en imagen potenciada en T2, los espacios perivasculares de la sustancia gris profunda se destacan en un patrón cribiforme
- Las gangliosidosis son enfermedades que manifiestan anomalías de la sustancia blanca y atrofia cortical (cuadro 7-6). Pueden ocurrir infartos lacunares y de sustancia blanca de vasos pequeños en la enfermedad de Fabry y Gaucher. De las lipidosis, la ataxia cerebelosa y la atrofia cerebelosa son más comunes en Enfermedad de Tay-Sachs y enfermedad de Niemann-Pick. Las anomalías talámicas bilaterales con hiperdensidad en la TC son características de la enfermedad de Sandhoff, pero también se pueden observar con enfermedad de Sandhoff, enfermedad de Tay-Sachs, enfermedad de Fabry y exposiciones tóxicas (fig. 7-38)
- FIGURA 7-38 Enfermedad de Fabry. A, La TAC sin contraste muestra calcificación aislada en el región ulvinar de los tálmos (flechas), que se considera un rasgo de imagen característico de esta afección. B, Más superiormente, los cambios isquémicos crónicos están presentes en la sustancia blanca periventricular.
- FIGURA: RMN T2 axial en niño a los 3 meses de edad (A), 8 meses (B) y 2 años de edad (C) muestran alta intensidad de señal bilateral simétrica putaminal y del caudado, con pérdida progresiva de volumen de estas estructuras a lo largo del tiempo. La enfermedad de Leigh puede ser el prototipo de los desórdenes de mitocondriales enzimáticos. Se cree que la enfermedad se debe a una deficiencia en las enzimas asociadas con el desglose de piruvato; su acumulación conduce a la acumulación de ácido láctico. Se manifiesta clínicamente por anomalías del sistema motor, ataxia, nistagmo, oftalmoplejía, espasticidad, retraso psicomotor, parálisis craneales y acidosis metabólica. La enfermedad de Leigh es un trastorno neurodegenerativo observado en niños menores de 2 años y expresada en RM por intensidad de señal anormalmente alta de la putamina bilateral en T2WI (fig. 7-39). Otras áreas también pueden mostrar una intensidad anormal, incluidos los núcleos caudados, el globo pálido, tálamo y tronco cerebral. Afectación del tronco encefálico inferior se correlaciona con la pérdida del control respiratorio, un potencial complicación fatal de la enfermedad. Supratentorial difusa La hiperintensidad T2 de la sustancia blanca puede acompañar a la profunda Hallazgos de materia gris. Estas áreas no se realzan. el señor puede también detectar atrofia del mesencéfalo y de la médula espinal en este trastorno. El hallazgo de laboratorio revelador es la acidosis metabólica con niveles elevados de lactato; el lactato puede detectarse con espectroscopia de RM de protones o fósforo examen.
- El síndrome MELAS representa una forma inusual de dismetabolismo mitocondrial. Los estudios de imágenes demuestran múltiples áreas de anormalidad cortical de alta intensidad de señal en T2WI, que a menudo cruza la distribución de una arteria cerebral tradicional (fig. 7-40). Aunque las lesiones pueden ser brillantes en DWI exploraciones, por lo general NO muestran valores de coeficiente de difusión aparente (ADC) reducidos y, por lo tanto, no son infartos agudos Los pacientes pueden tener ceguera cortical. Curiosamente, estas lesiones pueden desaparecer con el tiempo, dejando sólo una mínima dilatación del surco en su lugar. La anormalidad se asocia con evidencia serológica y espectroscópica. de niveles elevados de ácido láctico. Las lesiones asociadas a También se han reportado MELAS en putamen, caudado núcleos y tálamos. Puede haber realce de contraste. Aunque la resolución de las lesiones es el curso esperado, uno puede ver aparecer nuevas lesiones también. Por último, se espera una atrofia que favorezca la parte posterior del cerebro.
- FIGURA 7-40 Síndrome de MELAS. A, Imagen axial ponderada por difusión que muestra una hiperintensidad de difusión de base cortical en el lóbulo occipital izquierdo y temporal posterior. B, recuperación de inversión atenuada por líquido axial (FLAIR) y (C) imagen axial ponderada en T2 alta intensidad de señal en la corteza inflamada en la región de anomalía de difusión. Tenga en cuenta que la anormalidad es no se limita a un solo territorio vascular, sino que se extiende por la arteria cerebral media derecha y la arteria cerebral posterior. distribuciones arteriales sin afectar los ganglios basales.
- FIGURA: RM T2WI axial de un adolescente que demuestra del globo hiperintensidad del globo palido bilateralmente (flecha gorda) y de los tractos corticoespinales (flecha). Nota la pérdida sutil de arborización de mielina en el fibras en U subcorticales (flecha subcortical) Otras encefalomiopatías mitocondriales incluyen Síndrome de Kearns-Sayre (que también afecta a los ganglios basales) y síndrome MERRF. MERRF tiene una tasa más alta de lesiones focales del tronco encefálico que MELAS o Kearnes-Sayre. El síndrome de Kearns-Sayre afecta las órbitas con retinitis pigmentosa, oftalmoplejía, debilidad de los músculos extraoculares (y posible atrofia), y afecta el corazón, lo que resulta en los déficits de conducción cardiaca y una miocardiopatía. la calcificación de los ganglios Basales se produce en el síndrome de Kearns-Sayre con más frecuencia que en el síndrome MELAS o MERRF. Los ganglios basales hiperintensos, la atrofia cerebral y cerebelosa y la hiperintensidad difusa de la sustancia blanca son los más frecuentes. hallazgos comunes de RM en el síndrome de Kearns-Sayre. También pueden ocurrir microcefalia, hipoplasia cerebelosa y desmielinización de la sustancia blanca.
- FIGURA: RM coronal en T2WI en un paciente de 2 días de vida, donde se observa agrandamiento ventricular, un quiste germinolítico en el cuerno frontal derecho (flecha negra) , microgyria difusa cerebral y cerebelosa (flechas curvas), y aumento de la señal anormal en la materia blanca profunda
- los trastornos metabólicos de aminoácidos (cuadro 7-8) se ven generalmente en niños y se manifiestan neurológicamente por retraso en el desarrollo, retraso mental, convulsiones, y vómitos (cuando no se diagnostican temprano mediante exámenes de detección) pruebas). La maduración normal del cerebro a menudo se retrasa, y la mielinización puede verse afectada. La atrofia cerebral se ve en las últimas etapas. Por lo general, las manipulaciones dietéticas y/o las terapias de reemplazo son efectivas para obtener algún retorno de funcionamiento y control de la enfermedad.
- FIGURA: RM T2WI axial en un niño de 8 meses con enfermedad de Krabb, se observan a nivel de los nucleos del cerebelo, aonas hiperintensas correspondientes a edema (flecha recta) e hipointesas (núcleos del cerebelo, flecha curva), e hiperintenso en la sustancia blanca del cerebelo (flecha gorda). se debe a la deficiencia de galactocerebrósido-beta galactocerebrosidasa. El gen de esta enfermedad. se ha mapeado en el cromosoma 14. Los cerebrosidos de la mielina catabolizada no se puede degradar a galactosa y ceramida en esta condición y conduce a una acumulación dañina de galactosilesfingosina cerebrosido y psicosina en el parénquima cerebral. Cuando la enfermedad se presenta antes edad 2 (la forma infantil), los tractos piramidales, cerebeloso sustancia blanca, sustancia gris profunda, cuerpo calloso posterior, y la sustancia blanca parietooccipital suelen estar involucradas (ya que son las primeras zonas en mielinizarse). Las fibras en U son generalmente a salvo. Puede verse hiperdensidad de los tálamos en TC. Los hallazgos de la MRS incluyen colina y mioinositol anormalmente elevados (que se cree que son secundarios a la mielina). productos de descomposición o metabolismo de la membrana de fosfolípidos). Hay una marcada disminución en la anisotropía relativa de los tractos de materia blanca afectados identificados en exploraciones ponderadas por difusión. La atrofia finalmente se desarrolla. En el grupo de inicio tardío, pueden estar involucradas las mismas ubicaciones. a excepción de la sustancia blanca del cerebelo y la sustancia gris profunda. El tracto corticoespinal está involucrado constantemente (como lo está en el forma infantil). La enfermedad esplénica también es común en esta forma. El tracto motor superior, correspondiente a la extremidad inferior. región, se ve afectada en mayor medida que las regiones que subserve la cara y los brazos. La atrofia cerebral está presente en enfermedad temprana pero no tardía. La atrofia óptica está presente en ambos variedades Los ensayos de tratamiento de trasplante de células madre han sido alentador. El tratamiento exitoso ha correspondido con una tendencia hacia una mayor anisotropía relativa.
- FIGURA: T2WI axial en un bebé de 7 meses revela fisuras de sylvio agrandado (flecha gorda). Tenga en cuenta el edema y aumento anormal de señal (hiperintensidad) de los ganglios de la base (flecha curva) : incluyendo el cabezas de núcleos caudados, las putamina, y el globi pallidi bilateralmente. La acidemia glutárica puede sugerirse cuando uno encuentra macrocefalia con opérculos abiertos y anormalmente alta señal en los ganglios basales, especialmente en el globo pálido. Los pacientes pueden presentar encefalitis. Otras imágenes los hallazgos incluyen atrofia, dilatación del espacio subaracnoideo anterior a los lóbulos temporales y el volumen de los ganglios basales pérdida. En algunos pacientes pueden ocurrir anomalías en el sangrado. con esta afección y hemorragias intracraneales (a menudo SDH) de edad variable se pueden apreciar en las imágenes. Eso Puede ser difícil distinguir las hemorragias subdurales en esta afección de las que ocurren en el contexto de un traumatismo no accidental (lesión infligida).
- FIGURA: ultrasonido transfontanelar, corte parasagital de un recién nacido sintomático con enfermedad de la orina de jarabe de maple, que demuestra aumentado notable de la ecogenicidad de los tálamos debido al edema severo. En la enfermedad de la orina de jarabe de mapple (secundaria a anomalías descarboxilación de aminoácidos de cadena ramificada), cabeza la ecografía puede mostrar un aumento simétrico de la ecogenicidad de sustancia blanca periventricular, ganglios basales (principalmente pallidi) y tálamos. La TC y la RM muestran edema difuso en lugares similares, así como en el cerebelo y la cápsula. regiones. La mielinización retardada está presente. MRS puede mostrar una pico a 0,9 ppm que representa el amino de cadena ramificada picos de ácido
- La homocistinuria es causada por la deficiencia de cistationina beta sintasa, mapeada en el cromosoma 21. Los altos niveles plasmáticos resultantes de homocisteína dan como resultado múltiples eventos trombóticos, incluidos los accidentes cerebrovasculares. Se produce ateroesclerosis prematura. Las trombosis pueden ser arteriales o venoso. La presencia de subluxaciones del cristalino podría sugerir un diagnóstico de síndrome de Marfan, pero aquellos con homocistinuria están "arriba y adentro" y aquellos con enfermedad de Marfan están "abajo y afuera". Otros hallazgos neurorradiológicos pueden incluyen atrofia óptica, osteoporosis, cataratas, escoliosis, y cuerpos vertebrales bicóncavos.
- FIGURA: Hallazgo de neuroimagen en MMA: Niño varón con MMA (defecto cblA). Se obtuvo un estudio de resonancia magnética a la edad de 6 meses debido a irritabilidad, dificultades de alimentación, retraso en el desarrollo, encefalopatía y descompensación metabólica. Los núcleos caudado y lenticular están edematosos e hiperintensos en la imagen axial turbo spin-eco (SE) ponderada en T2 (W) (A) Acidemia propiónica (APP) y acidemia metilmalónica (MMA) son trastornos hereditarios del ciclo tricarboxílico causado por defectos enzimáticos, que pueden conducir a alteraciones metabólicas acidosis. Estas enfermedades se detectan dentro del primer mes de vida ya que los bebés tienen dificultades para alimentarse, hipotonía muscular, coreoatetosis, microcefalia y convulsiones. La enfermedad ocurre con más frecuencia en pacientes de Arabia Saudita donde se transmite como un trastorno autosómico recesivo (aunque también se puede ver con deficiencia de biotina y/o cobalamina). Atrofia y señal anormal de sustancia blanca con retraso mielinización se ven en la resonancia magnética convencional. ganglionar basal lesiones (típicamente en el globo pálido en MMA y en el globo pálido, caudados y putamina en PPA) después de 1 año de puede aparecer la edad. La etiología no está clara y, a veces, resolver. Pueden verse picos de lactato en la MRS de protones. Estos dos entidades se parecen excepto que la materia blanca de baja densidad en CT está presente en PPA pero no en MMA. Los retrasos en la mielinización son más evidentes en MMA.
- FIGURA: Imagen por resonancia magnética de cráneo. Corte axial en secuencia FLAIR que revela atrofi a difusa córtico y subcortical, así como aumento del espacio subaracnoideo y atrofia cerebelosa. La lipofuscinosis ceroide neuronal juvenil es un trastorno neurodegenerativo lisosomal causado por la acumulación de lipopigmento en neuronas. La forma neonatal de esta enfermedad. se asocia con microcefalia y déficits visuales. Pérdida de volumen del SNC, más prominente en el cerebelo, tálamo hipointenso y periventricular hiperintenso sustancia blanca en T2WI y espectros MRS de protones que revelan reducción de NAA y aumento de mioinositol y glutamato/ glutamina caracterizan este trastorno. Los anillos periventriculares de alta señal se observan después de 1 año de vida. Hipointensidad de la También se observan tálamos y ganglios basales e hiperintensidades similares al LCR peritrigonal en exploraciones T2WI
- Comprender el desarrollo de el sistema nervioso central (SNC) ayuda a comprender anomalías congénitas del cerebro y la columna vertebral y la coexistencia de múltiples anomalías. Aunque hay cierta superposición, para poner simplemente, el SNC pasa por seis eventos principales de desarrollo que incluyen (1) neurulación primaria (3 a 4 semanas de gestación); (2) desarrollo promesencefálico (2 a 3 meses de gestación); (3) proliferación neuronal (3 hasta los 4 meses de gestación); (4) migración neuronal (3 a 5 meses de gestación); (5) organización (5 meses de gestación a años postnatales); y (6) mielinización (desde el nacimiento hasta los años posteriores al nacimiento). Los períodos de terminación mencionados anteriormente de cada evento significa que la malformación de ese evento en particular puede tener su inicio antes de que termine el event
- La neurulación es una serie de eventos inductivos que tienen lugar en la cara dorsal del embrión y da como resultado el desarrollo del cerebro y la médula espinal. neurulación primaria involucra eventos que están relacionados con la formación del cerebro y médula espinal exclusiva de su región más caudal. Él La primera fusión de los pliegues neurales se produce en la parte inferior del bulbo raquídeo. El cierre generalmente procede rostral y caudalmente, aunque no es un simple proceso similar a una cremallera. Trastornos de primaria neurulación son craneosquisis, anencefalia, mielosquisis, encefalocele, mielomeningocele y malformaciones de Chiari II. Varias malformaciones del cierre del tubo neural son acompañada de anomalías del esqueleto axial, meningovasculares y de la cubierta dérmica. La neurulación secundaria ocurre más tarde que la neurulación primaria con procesos secuenciales de canalización y diferenciación regresiva. Los trastornos de la neurulación secundaria dan como resultado malformaciones del sacro inferior y segmentos coxígeos del tubo neural que afectan al cono medular, filum terminale y cauda equina. lipomas de el filum, fila atada corta y engrosada y cuerdas atadas, y el síndrome de regresión caudal con agenesia sacra son también figuran en esta categoría.
- La relación inductiva principal se da entre el mesodermo precordal y el mesodermo de la cuerda noto y el prosencéfalo, y toma lugar ventralmente en el extremo rostral del embrión, por lo tanto algunos autores utilizan el término inducción ventral. Durante este evento se produce la formación de la cara y el cerebro anterior, las malformaciones del cerebro suelen ir acompañadas de anomalías faciales (como ciclopía y probóscide). El espectro de malformaciones puede ser tan grave como la aprosencefalia, a quizás malformaciones callosas clínicamente ocultas. Tres Los principales eventos secuenciales (y malformaciones relacionadas) son desarrollo promesencefálico (atelencefalia), hendidura encefálica premes (holoprosencefalia) y línea media desarrollo promesencefálico (agenesia del cuerpo calloso, agenesia del septum pellucidum, displasia septoóptica, displasia hipotalámica).
- MIGRACIÓN NEURONAL: Las neuronas migran desde las zonas ventricular y subventricular del subepéndimo a su residencia final para la vida. Inicialmente, las neuronas migran por translocación de la célula. seguido de dos variedades básicas de migración celular: radial y tangencial. La migración radial conduce a la proyección. neuronas de la corteza cerebral y núcleos profundos en el cerebro y células de Purkinje en el cerebelo. Tangencial La migración conduce a las interneuronas de la corteza cerebral y capa granular interna de la corteza cerebelosa. Las capas de las neuronas están al revés: las neuronas que llegan temprano son profundas, mientras que las neuronas que llegan tarde están en el aspecto superficial de la corteza. Los trastornos de la migración neuronal incluyen esquizencefalia, lisencefalia (paquigiria), polimicrogiria, heterotopía y disgenesia cerebrocortical focal.