



![[F]
EXTERIOR
INTERIOR
[F]
[F] [F] DISTRIBUCIÓN
EFECTO
Tejido susceptible
FIJACIÓN
METABOLISMO
EXCRECIÓN
iv
im
oral](https://image.slidesharecdn.com/biocineticabiotrans10mar-130607092947-phpapp02/85/Biocinetica-biotrans-10mar-5-320.jpg)

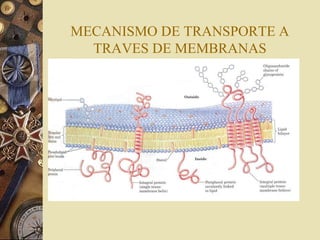

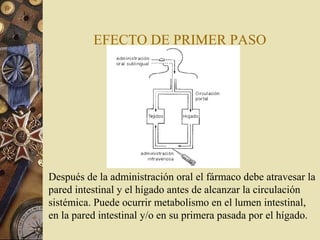

















































Este documento trata sobre la farmacocinética y biotransformación de fármacos. Explica conceptos como la absorción, distribución, metabolismo y excreción de medicamentos en el organismo, así como los factores que afectan estos procesos. También describe las vías de administración de fármacos y los mecanismos de transporte a través de membranas, así como las fases del metabolismo y los factores que influyen en él.





























































