Descargado 46 veces

![1188 Farmacología humana
y, por lo tanto, tienen un espectro restringido de actividad
antiviral; además, prácticamente ninguno carece de toxi- O O O
II II II
cidad. Asimismo, debido a su mecanismo de acción, no son N N N
HN HN HN
efectivos frente al virus que no esté replicándose (virus la-
tentes); son fármacos virostáticos. H2N N N H 2N N N H2N N N
Por último, la aparición de la epidemia del sida ha in-
O HOCH2 O HOCH2 O
tensificado la búsqueda de nuevos agentes antivirales y HOCH2
aunque la terapia antiviral en general se beneficia de este H2C CH2 HOH2C–CH CH2
esfuerzo, la mayor parte de los nuevos agentes son anti-
rretrovirales. Por ello se dividirá este capítulo en dos gran-
OH
des apartados: agentes para infecciones por virus que no
son los de la inmunodeficiencia humana (antivíricos no Desoxiguanosina Aciclovir Ganciclovir
VIH) y agentes para infecciones por VIH.
NH2 O
I II
N H2N–C N
N
II. ANTIVÍRICOS NO VIH N
N
N N
HOCH2 O HOCH2 O
A. ANÁLOGOS DE LOS NUCLEÓSIDOS
OH
1. Aciclovir
OH OH OH
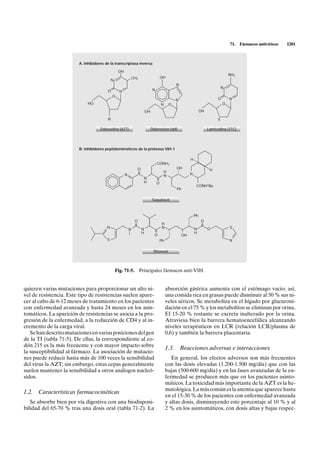
El aciclovir (acicloguanosina, 9-[2-hidroxietoximetil]-
guanina), análogo acíclico del nucleósido natural 2´-de- Vidarabina Ribavirina
soxiguanosina, tiene una potente acción antiviral frente NH2
a muchos Herpesvirus (fig. 71-1). OH
I O
HO – P – C
II
1.1. Actividad antivírica, mecanismo de acción O
OH
y resistencias
Amantadina Foscanet
Es especialmente activo frente al virus del herpes sim-
ple (VHS) de los tipos 1 y 2 (CI50 0,1-1,6 µM) y el virus
de la varicela zoster (VVZ) (CI50: 3-4 µM); en orden des- Fig. 71-1. Estructura de fármacos antivíricos no-VIH.
cendente también presenta actividad in vitro frente al
virus de Epstein-Barr (VEB), virus del herpes humano
de tipo 6 (VHH-6) y citomegalovirus (CMV). Frente al Dado que el crecimiento celular sólo llega a inhibirse
CMV, su actividad es mucho menor que la de ganciclo- a concentraciones muy altas de aciclovir (> 300 µM), el
vir, foscarnet y vidarabina; muchas cepas son resistentes índice terapéutico de este fármaco es muy favorable
al aciclovir (CI50 > 200 µM). Su acción antiviral se mani- (> 3.000).
fiesta únicamente en virus en fase de replicación. Esta ac- La resistencia al aciclovir puede aparecer por diferen-
ción selectiva se debe al hecho de que en su primera fos- tes vías: a) el mecanismo más común es la aparición de
forilación a aciclovir-monofosfato interviene una enzima una mutación que genere una cepa deficiente en timidín-
propia del virus, la timidín-cinasa (no presente en el cinasa; b) aparición de una mutación que genere una ti-
CMV) (fig. 71-2). En células no infectadas por virus, esta midín-cinasa que no reconozca al aciclovir como sustrato,
primera fosforilación es muy lenta. De hecho, esta en- y c) la última conocida es la aparición de una mutación
zima viral tiene una afinidad por el aciclovir 200 veces su- que altere la sensibilidad de ADN-polimerasa viral al aci-
perior a la de la timidín-cinasa de la célula. clovir-trifosfato. En caso de resistencia al aciclovir se ha
Las posteriores fosforilaciones hasta alcanzar la forma recomendado el uso de foscarnet.
activa del fármaco, aciclovir-trifosfato, se llevan a cabo
mediante enzimas celulares. El aciclovir-trifosfato es ca-
1.2. Características farmacocinéticas
paz de inhibir la replicación viral por tres vías: a) inhi-
biendo selectivamente la ADN-polimerasa viral; b) me- La absorción oral es lenta y variable con una biodispo-
diante la competencia del aciclovir-trifosfato con la nibilidad del 15-30 %, alcanzando la concentración má-
guanosín-trifosfato por incorporarse al ADN viral, y c) xima plasmática a las 1,5-2,5 horas de la dosis (tabla 71-2).
actuando como finalizador de cadena al incorporarse al Por vía intravenosa se alcanzan concentraciones hasta
ADN viral. No se descarta que existan mecanismos adi- 10 veces superiores, mientras que tras la administración
cionales de acción. tópica no se detectan concentraciones plasmáticas, aun-](https://image.slidesharecdn.com/capitulo71-110426152826-phpapp01/85/Capitulo-71-2-320.jpg)






















Este documento describe los fármacos antivirales no anti-VIH. Explica que el aciclovir es potente contra el herpes simple y varicela-zoster. Su mecanismo de acción involucra fosforilación por timidina kinasa viral para convertirse en la forma activa trifosfato, que inhibe la ADN polimerasa viral. También cubre otros análogos de nucleósidos como ganciclovir, ribavirina y foscarnet, así como sus mecanismos de acción y espectros de actividad.






![Antivirales[1]](https://cdn.slidesharecdn.com/ss_thumbnails/antivirales1-171006005651-thumbnail.jpg?width=640&height=640&fit=bounds)








































