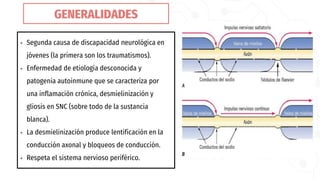
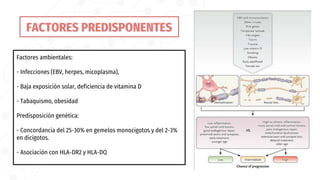
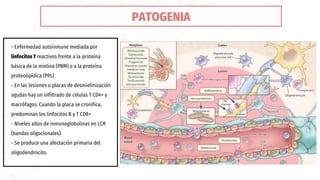
La esclerosis múltiple es la segunda causa de discapacidad neurológica en jóvenes. Se caracteriza por una inflamación crónica y desmielinización en el sistema nervioso central, causando lentificación en la conducción nerviosa. Afecta principalmente a mujeres jóvenes de raza blanca y del norte de Europa. Su diagnóstico requiere evidencia de lesiones diseminadas en el tiempo y espacio mediante resonancia magnética, y el descarte de otras enfermedades. Su tratamiento incluye corticoides para los brotes y