Descargado 203 veces



















![ESCLEROSIS MULTIPLE
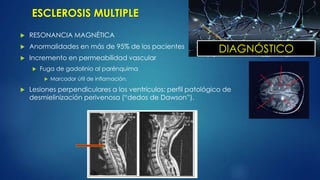
LCR
Pleocitosis de mononucleares
Mayor [] de IgG sintetizada en el LCR
Proteínas incrementadas o nls.
Medición de bandas oligoclonales: valora la producción
intrarraquídea de IgG.
Pleocitosis que excede de 75 cels/mcL, presencia e PMN o
proteínas >1g/L deben despertar la sospecha de otro
padecimiento.
DIAGNÓSTICO](https://image.slidesharecdn.com/patologiasneuromusculares-130623203455-phpapp02/85/Patologias-neuromusculares-21-320.jpg)








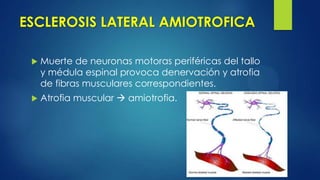




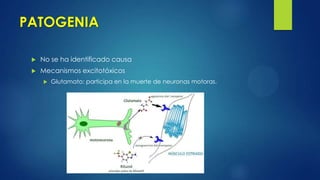
















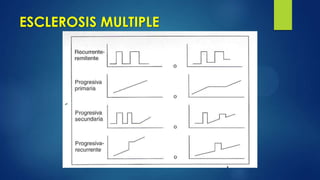
Este documento describe la esclerosis múltiple, una enfermedad desmielinizante del sistema nervioso central. Explica que causa inflamación y destrucción selectiva de la mielina, lo que interrumpe la conducción nerviosa. Describe los cuatro tipos de esclerosis múltiple, los criterios de diagnóstico, las manifestaciones clínicas, y los enfoques de tratamiento como terapias modificadoras de la enfermedad y tratamiento sintomático.


























































![ENFERMEDADES_DESMIELINIZANTES[1] UNDAC PASCO.pptx](https://cdn.slidesharecdn.com/ss_thumbnails/enfermedadesdesmielinizantes1-260120163022-ce8323fc-thumbnail.jpg?width=640&height=640&fit=bounds)


























