Descargado 24 veces


![Cinética del electrodo
No existe “caída” IR
No existe sobrepotencial
Existe IR-drop y sobrepoentiales η, V
d
iIR
Conductividad del electrolito κ [S/m]](https://image.slidesharecdn.com/introduccionalaelectroquimicap2-150518044002-lva1-app6892/85/Introduccion-a-la-electroquimica-p2-3-320.jpg)


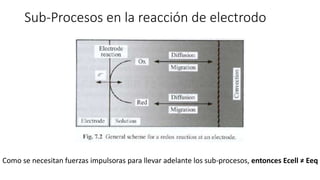
![Densidad de corriente y sobrepotencial
• Velocidad de reacción química heterogénea: J [mol m-2 s-1]
También es valida para reacciones electroquímicas!
• Densidad de corriente: j = nFJ [(As mol-1)*(mol m-2 s-1)=A m-2]
• Como varia el voltaje de la celda y el potencial del electrodo con respecto a la densidad de
corriente?
• Sobrepotencial en el electrodo: (=Fuerza impulsora para el proceso)
• η y j son positivos para Corrientes anódicas y negativos para Corrientes catódicas
Potencialdeelectrodo
Corriente
Esquemáticamente tenemos:
rEE ](https://image.slidesharecdn.com/introduccionalaelectroquimicap2-150518044002-lva1-app6892/85/Introduccion-a-la-electroquimica-p2-7-320.jpg)









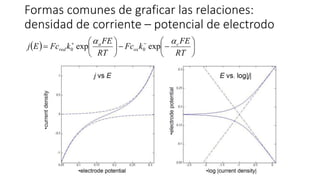


El documento presenta conceptos básicos de cinética electroquímica, incluyendo densidad de corriente, sobrepotencial, curvas de polarización, teoría de complejos activados y ecuaciones que relacionan el potencial y la densidad de corriente en un electrodo. Explica cómo la densidad de corriente depende exponencialmente del sobrepotencial según la ecuación de Butler-Volmer.















































![Teoriacorrosion[1]5577](https://cdn.slidesharecdn.com/ss_thumbnails/teoriacorrosion15577-101004200527-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)






































