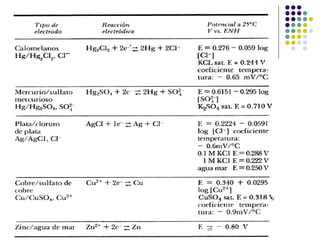
Descargado 681 veces


















![Si [productos] > [reactivos] ; Q > 1 y log Q > 0. El producto se resta de ΔE° y ΔE < ΔE°. Si [productos] < [reactivos] ; Q < 1 y log Q < 0 El producto se hace > 0 y ΔE > ΔE°. Potencial de celda y constante de equilibrio Si ΔG = 0 , el sistema está en equilibrio. como una fem = 0 significa que no se está produciendo una reacción neta en la celda voltaica, la reacción de la celda ha alcanzado el equilibrio. Cuando ΔE = 0, Q = K](https://image.slidesharecdn.com/electroqumicaindustrial-110222171416-phpapp01/85/Electroquimica-industrial-22-320.jpg)




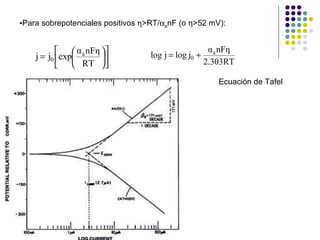
![La densidad de corriente observada a cada valor de sobrepotencial, , j depende de: La densidad de corriente de intercambio, j 0 . Los coeficientes de transferencia, a y c . La temperatura. Cuando la densidad de corriente puede medirse a sobrepotenciales positivos y negativos, conviene modificar la ecuación de Butler-Volmer para analizar mejor los datos. Así, si dividimos por exp( a nF /RT) la ecuación de Butler-Volmer y aceptando que c = 1- a , se obtiene : Así midiendo j y , se puede representar log [ ] frente a y debe ser una línea recta de donde se pueden determinar parámetros cinéticos de la ordenada en el origen, j 0 , y de la pendiente, a .](https://image.slidesharecdn.com/electroqumicaindustrial-110222171416-phpapp01/85/Electroquimica-industrial-27-320.jpg)






















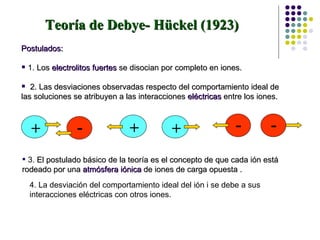
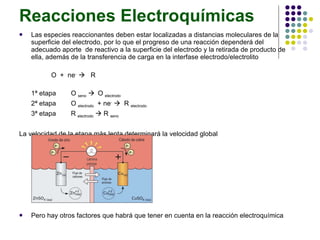
El documento introduce la ingeniería electroquímica, que involucra el diseño y operación de procesos que convierten energía química en eléctrica. Cubre conceptos como potencial de electrodo, voltaje, intensidad de corriente y densidad de corriente. También explica teorías como la de Debye-Hückel sobre la conductividad de electrolitos y tipos de reacciones electroquímicas.





















































