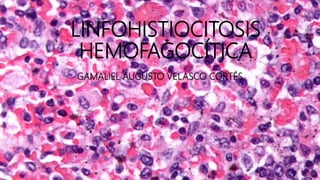
Este documento describe la linfohistiocitosis hemofagocítica (LHH), un síndrome de hiperinflamación que causa daño multisistémico. La LHH se caracteriza por una regulación inmune defectuosa que causa una actividad excesiva de linfocitos T y macrófagos, lo que conduce a una liberación masiva de citoquinas inflamatorias. El documento discute los criterios diagnósticos, las posibles causas, los síntomas clínicos, y el enfoque terapéutico que incl