Descargado 27 veces


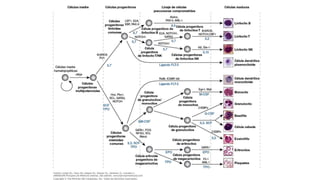





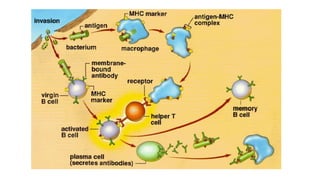









Este documento proporciona una introducción a la histiocitosis y la linfohistiocitosis hemofagocítica (HLH). Define la HLH como un síndrome de activación inmune patológica caracterizado por una proliferación incontrolada de macrófagos y linfocitos T citotóxicos. Explica que la HLH puede ser primaria debido a defectos genéticos o secundaria a infecciones, neoplasias u otras enfermedades. También resume los criterios de diagnóstico clínico y los tratamientos















































































