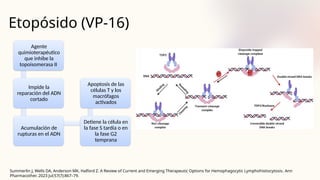
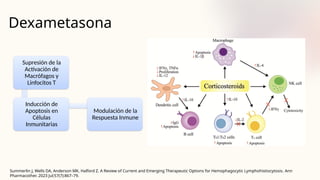
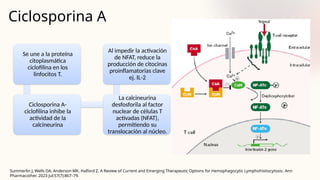
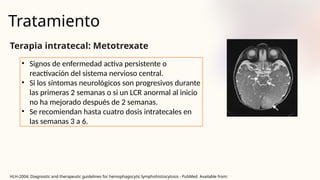
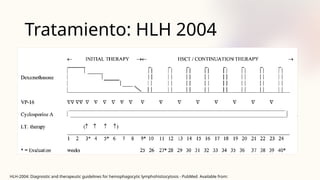
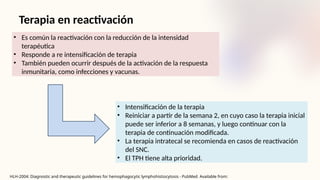
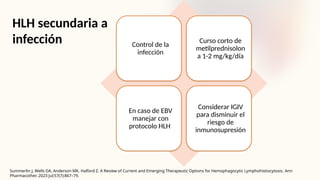
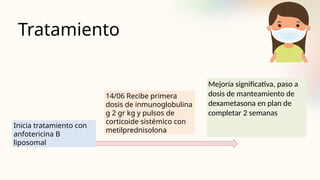
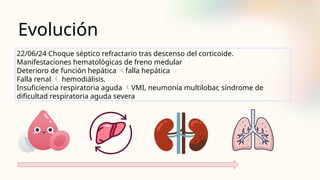
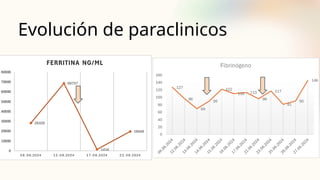
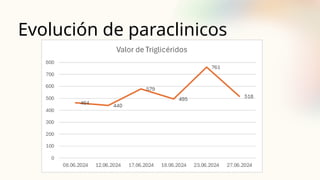
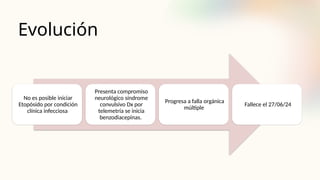
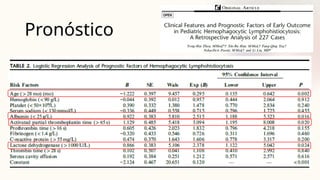
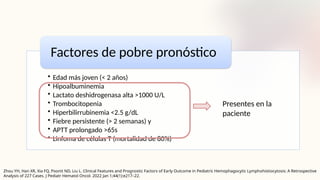
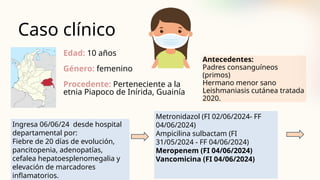
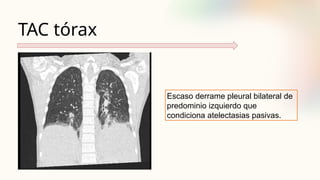
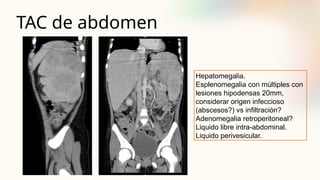
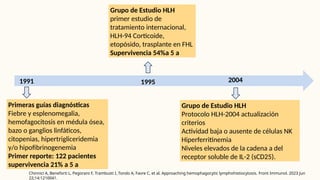
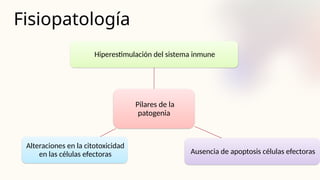
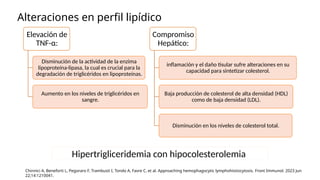
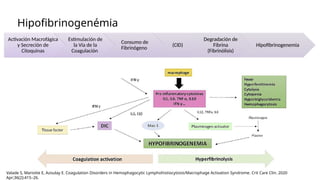
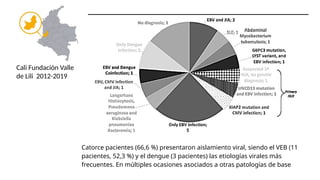
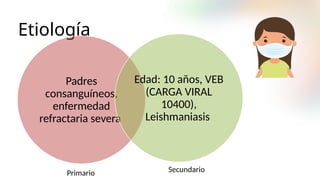
El documento describe un caso clínico de linfocitosis hemofagocítica en una niña de 10 años con antecedentes familiares relevantes y síntomas como fiebre, pancitopenia y hepatosplenomegalia. Se aborda la epidemiología, fisiopatología, diagnóstico y tratamiento de la enfermedad, resaltando la relación con infecciones como la leishmaniasis. Además, se presenta una revisión de la historia de la enfermedad, sus clasificaciones y características clínicas.




















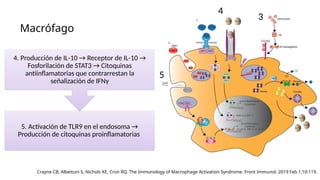








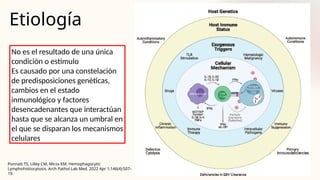
![Virus de Epstein Barr
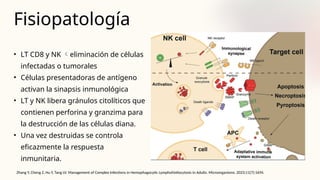
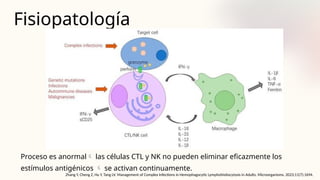
• Suele atacar a las células B, pero en un subgrupo de pacientes
con VEB‑HLH, predominantemente origen asiático, infecta a las
células T o NK, lo que conduce a una proliferación oligoclonal o
monoclonal y a una producción masiva de citocinas
• Agresivo con frecuencia afecta al sistema nervioso central.
Pediatric hemophagocytic lymphohistiocytosis | Blood | American Society of Hematology [Internet]. [cited 2024 Jul 21]. Available from:
https://ashpublications.org/blood/article/135/16/1332/452577/Pediatric-hemophagocytic-lymphohistiocytosis](https://image.slidesharecdn.com/hemofagocticoenpediatra-241210025807-d81bc264/85/sindrome-HEMOFAGOCITICO-EN-PEDIATRIA-pptx-43-320.jpg)
![Virus de
Epstein Barr
Pediatric hemophagocytic lymphohistiocytosis | Blood | American Society of Hematology [Internet]. [cited 2024 Jul 21]. Available from:
https://ashpublications.org/blood/article/135/16/1332/452577/Pediatric-hemophagocytic-lymphohistiocytosis](https://image.slidesharecdn.com/hemofagocticoenpediatra-241210025807-d81bc264/85/sindrome-HEMOFAGOCITICO-EN-PEDIATRIA-pptx-44-320.jpg)




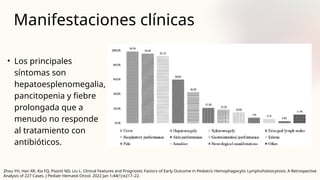








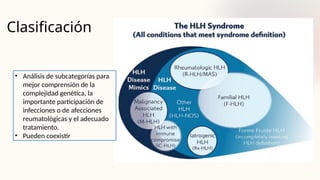
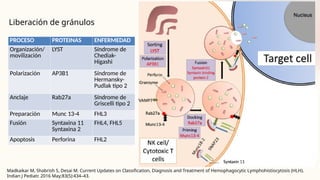
![Aproximación diagnóstica
Pediatric hemophagocytic lymphohistiocytosis | Blood | American Society of Hematology 2020. [cited 2024 Jul 21]. Available from:
https://ashpublications.org/blood/article/135/16/1332/452577/Pediatric-hemophagocytic-lymphohistiocytosis](https://image.slidesharecdn.com/hemofagocticoenpediatra-241210025807-d81bc264/85/sindrome-HEMOFAGOCITICO-EN-PEDIATRIA-pptx-64-320.jpg)
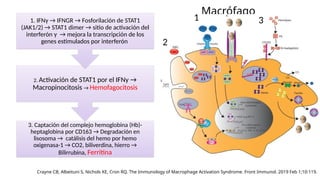
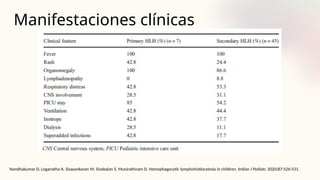
![Aproximación diagnóstica
Pediatric hemophagocytic lymphohistiocytosis | Blood | American Society of Hematology 2020. [cited 2024 Jul 21]. Available from:
https://ashpublications.org/blood/article/135/16/1332/452577/Pediatric-hemophagocytic-lymphohistiocytosis](https://image.slidesharecdn.com/hemofagocticoenpediatra-241210025807-d81bc264/85/sindrome-HEMOFAGOCITICO-EN-PEDIATRIA-pptx-65-320.jpg)