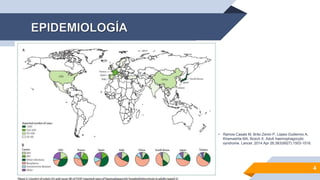
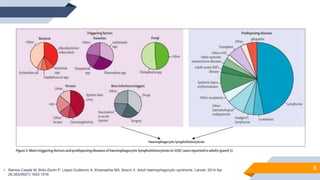
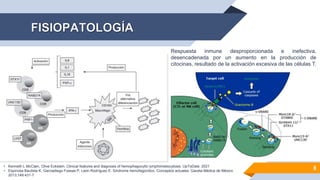
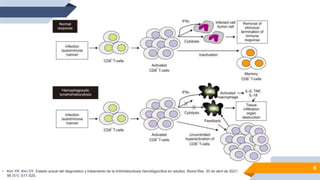
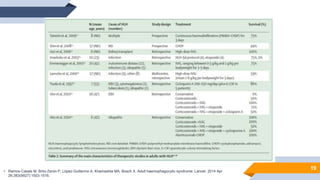
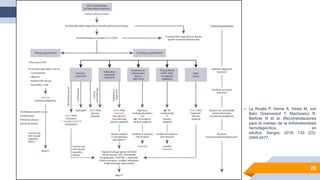
El documento describe el síndrome hemofagocítico (SH), una enfermedad rara causada por una respuesta inmune desproporcionada e inefectiva. Puede ser hereditaria o adquirida, asociada con infecciones, neoplasias u otras enfermedades. Los síntomas incluyen fiebre, baja en glóbulos blancos y plaquetas, y elevación de marcadores inflamatorios. El diagnóstico se basa en criterios clínicos y de laboratorio. El tratamiento busca suprimir la hiperinflam