Descargar como PDF, PPTX


![MANUAL DE
PREVENCIÓN Y
TRATAMIENTO DE
RIESGOS DE
DISCAPACIDADES
EN PERI-
NEONATOLOGÍA
[Con los Criterios de la CIF-
OMS]
[La patología perineonatal es causa de trastornos severos
que provocarían lesiones discapacitantes, susceptibles de
prevención y tratamiento]
2014
MINISTERIO DE SALUD PÚBLICA
DEL ECUADOR
Dirección de Discapacidades](https://image.slidesharecdn.com/patologiaperineonatal2-150209213016-conversion-gate02/85/Patologia-perineonatal2-2-320.jpg)
![[La patología perineonatal
es causa de trastornos
severos que provocarían
lesiones discapacitantes,
susceptibles de prevención
y tratamiento]](https://image.slidesharecdn.com/patologiaperineonatal2-150209213016-conversion-gate02/85/Patologia-perineonatal2-3-320.jpg)






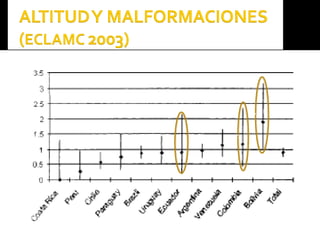
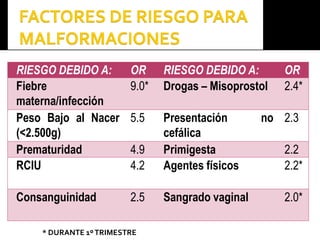
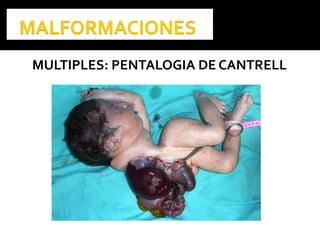
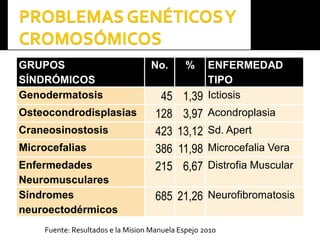

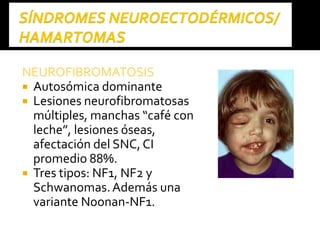



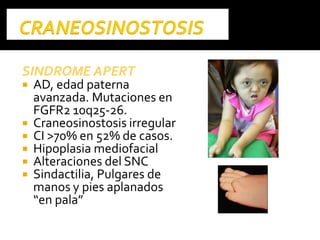
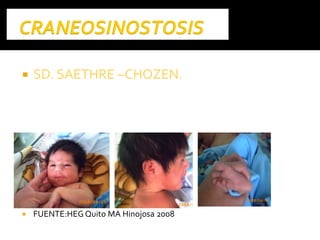

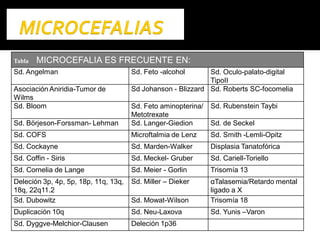

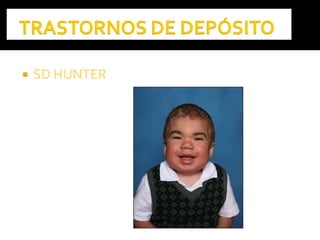


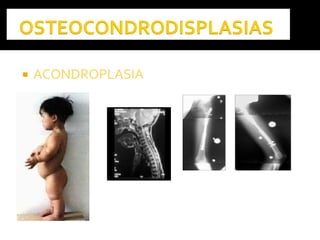
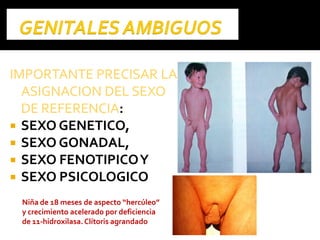
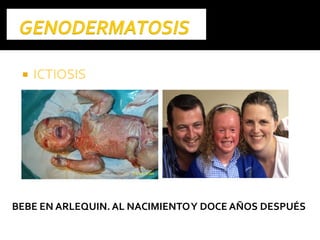
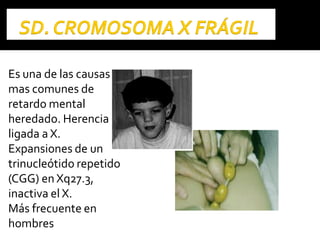
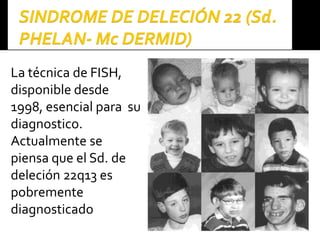

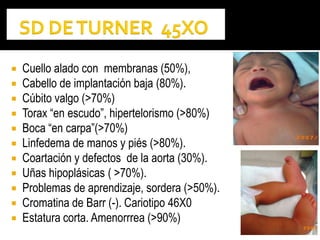
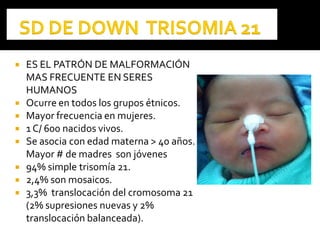
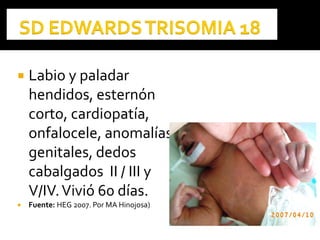

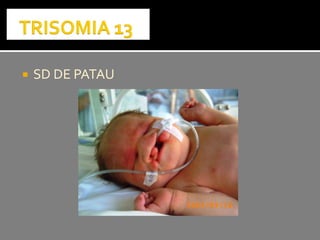
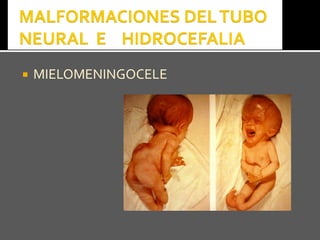

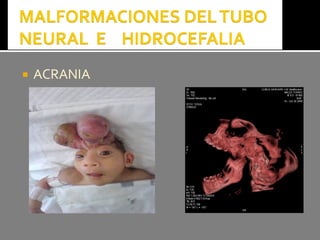
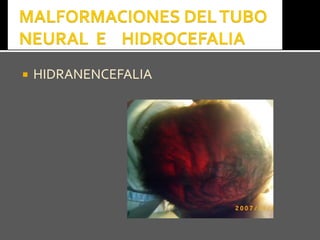




Este documento presenta información sobre la prevención y tratamiento de riesgos de discapacidades en neonatología. Describe las causas de patologías perinatales que pueden provocar discapacidades, incluyendo infecciones, defectos genéticos y ambientales. Proporciona datos sobre la prevalencia de discapacidades en Ecuador y los costos asociados durante toda la vida. También describe varios síndromes y condiciones que pueden resultar en discapacidad si no son tratados adecuadamente durante el período perinatal.



































































