
Recomendados
Más contenido relacionado
La actualidad más candente
La actualidad más candente (20)
Destacado
Destacado (20)
Similar a Principios de la termodinámica
Similar a Principios de la termodinámica (20)
Último
Último (20)
Principios de la termodinámica
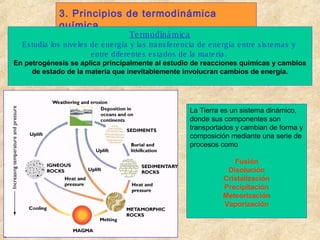
- 1. 3. Principios de termodinámica química Te rmodiná mica Es tudia los nive le s de e ne rgía y la s tra ns fe re ncia de e ne rgía e ntre s is te ma s y e ntre dife re nte s e s ta dos de la ma te ria . Termodinámica En petrogénesis se aplica principalmente al estudio de reacciones químicas y cambios de estado de la materia que inevitablemente involucran cambios de energía. La Tierra es un sistema dinámico, donde sus componentes son transportados y cambian de forma y composición mediante una serie de procesos como Fusión Disolución Cristalización Precipitación Meteorización Vaporización
- 2. Los métodos de termodinámica química emplean para Predecir la manera en que los sistemas de formación de rocas responden a cambios en presión, temperatura y composición química. Interpretar la P, T y composición química de formación de sistemas antiguos a partir de la composición química de rocas, minerales, vidrio, etc. Por lo menos en principio, podemos emplear la termo- dinámica para predecir: A que temperatura fundirá una roca y que composición tendrá el líquido generado. La secuencia de minerales que cristalizará para formar una roca ígnea a partir de un líquido. Los minerales nuevos que se formarán cuando esa roca se metamorfize. Los minerales y la composición de las soluciones que se forman cuando una roca se intemperiza La te rmodiná mica s ola me nte e s útil cua ndo s e a plica a s is te ma s e n e quilibrio. Si un sistema en equilibrio es perturbado, la termodinámica puede predecir el nuevo estado de equilibrio, pero no puede predecir como, que tan rápido o si se alcanzará ese estado de equilibrio.
- 3. Definiciones SISTEMA Cualquier parte del universo que se desea estudiar. La posición exacta de las fronteras del sistema se fija de acuerdo al problema que se desea estudiar. Un sistema puede ser por ejemplo un grupo de átomos, de minerales o de rocas. Los cambios que ocurren en el sistema pueden o no involucrar interacción con el entorno. S is te ma a is la do Tienen energía y masa constante debido a que sus fronteras son - rígidas, por lo que no permiten el intercambio de energía mecánica; - perfectamente aisladas, impidiendo el flujo de calor; - impermeables al intercambio de materia. Estos sistemas no ocurren en la naturaleza, sin embargo son importantes porque las reacciones ocurren en sistemas aislados son aquellas que no pueden liberar o absorber calor o cualquier otra forma de energía.
- 4. S is te ma ce rra do Transferencia de energía hacia dentro o hacia afuera del sistema; no hay inetercambio de materia. Tienen masa y composición constante, pero niveles de energía variables. Como el intercambio de materia es un proceso lento, procesos que ocurren en corto tiempo (p. ej. enfriamiento de un dique) se pueden considerar como sistemas cerrados. S is te ma a bie rto Transferencia tanto de energía como de materia hacia dentro o hacia afuera del sistema. El sistema puede estar abierto a una especie o varias especies químicas. La mayoría de los sistemas geológicos son abiertos, al menos en el contexto de la larga duración que pueden tener. S is te ma a dia bá tico Categoría especial de los sistemas aislados en el cual no se puede intercambiar calor con el entorno, pero se puede transferir energía a través de las fronteras del sistema en forma de trabajo. Una pluma del manto o un cuerpo de magma que asciende y se decomprime, se enfría mientras que se expande hacia el entorno y realiza un trabajo expansivo (P∆V). Como la tasa de conducción de calor es baja, muy poco calor es conducido al entorno.
- 5. FASE Cuerpo homogéneo de materia (sólido, líquido o gas) con fronteras definidas hacia otras fases, y que puede ser separado mecánicamente de las otras fases. Un sistema puede estar compuesto por una fase (sistema homogéneo) o por 2 o más fases (sistema heterogéneo). P. ej. todos los cristales de olivino en una roca constituyen la fase olivino; una solución de sal o una mezcla de gases constituyen una sola fase. COMPONENTE Describen la composición química de un sistema. Se define como el conjunto menor de fórmulas químicas necesarias Cuáles serían los para describir la composición de todas las fases de un componentes de: sistema. Mezcla de hielo, agua y Fase: vapor de agua. Olivino: Solución sólida de (Mg, Fe)2SiO4 Feldespato Componentes: (K, Na, Ca) (Al,Si)2 Si2O8 Mg2SiO4 MgO Mg 2+ Fe2SiO4 FeO Fe2+ Clinopiroxeno SiO2 Si4+ (Ca, Na) (Mg,Fe2+,Al3+) Si2O6 O2- Es más común emplear las fórmulas de miembros extremos, debido a que se tienen datos termodinámicos para ellos.
- 6. EQUILIBRIO Un sistema está en equilibrio termodinámico cuando no se observa ningún cambio en sus propiedades termodinámicas a lo largo del tiempo. Este estado tiene dos atributos: 1. En un sistema en equilibrio ninguna de sus propiedades cambian con el tiempo. 2. Un sistema en equilibrio retornará a ese estado después de haber sido perturbado, esto es al cambiar ligeramente uno o más parámetros y regresarlos nuevamente a sus valores originales. Cualquier sistema que tenga gradientes de temperatura, presión o composición, tenderá a cambiar hasta eliminar esos gradientes. P. ej. Una lava emplazada en la superficie, no está en equilibrio con el aire que la rodea, y se enfriará. Equilibrio estable Nivel de energía más bajo. Reúne atributos de equilibrio. P. ej. grafito. Equilibrio metaestable Reúne los atributos de equilibrio, pero no tiene el nivel energético más bajo. Sólo si se supera la barrera de energía (energía de activación) se accederá al estado estable. P. ej. Diamante en la superficie de la tierra Analogía mecánica de un sistema químico. Sistema inestable 1. Reúne primer atributo de equilibrio, Los sistemas naturales tienden a estados pero no el segundo. de mínima energía. 2. No reúne ninguno de los atributos
- 7. La forma estable de una sustancia es diferente a diferentes condiciones. P. ej. 1) A +5ºC la forma estable de H2O es agua, y a –5ºC es hielo. 2) A alta P y T, la forma estable de C es diamante, a baja P y T es grafito. VARIABLES o PROPIEDADES DE ESTADO Los sistemas en equilibrio tienen propiedades medibles. Una propiedad es cualquier cantidad que tiene un valor fijo e invariable en un sistema en equilibrio. (p. ej., temperatura, densidad, índice de refracción). Estas variables caracterizan a los estados termodinámicos y dependen sólo del estado presente del sistema y no de la forma en que se alcanzó ese estado. P ropie da de s e xte ns iva s Son proporcionales a la cantidad de materia considerada y por lo tanto dependen del tamaño del sistema. Estas propiedades son aditivas; el valor del todo es igual a la suma de las partes. P. ej. volumen, masa, energía. P ropie da de s inte ns iva s Son independientes de la cantidad de materia (del tamaño del sistema). No son aditivas. P. ej., concentración, temperatura, presión. Incluye propiedades molares, como el volumen molar.
- 8. P ROCES OS Son aquellos que afectan a un sistema termodinámico al cambiar de un estado a otro (p. ej. una reacción química). La trayectoria seguida en el cambio entre estados no es materia de la termodinámica, sino de la cinética. Se reconocen dos tipos extremos e ideales de proceso termodinámico: P roce s o te rmodiná mico irre ve rs ible Cambio de un estado metaestable a un estado más estable de menor energía. Ejemplo: Conversión de vidrio metaestable a cristales bajo condiciones atmosféricas (devitrificación). La devitrificación ocurre espontáneamente en la dirección de menor energía. P roce s o te rmodiná mico re ve rs ible Cambio de un estado inicial estable a un estado final también estable, pasando por una secuencia continua de estados de equilibrio. En la naturaleza no existen procesos perfectamente reversibles, se emplean sólo como modelos termodinámicos.
- 9. P rime ra Le y de la Te rmodiná mica Le y de la cons e rva ción de la e ne rgía : La e ne rgía no s e cre a ni s e de s truye Ene rgía inte rna La energía total, ET , de un sistema puede descomponerse en energía de masa (Em), energía cinética (Ek), energía potencial (Ep), y energía interna (U): ET = E m + E k + E p + U La energía interna U considera la energía de las partículas que constituyen el sistema y sus interacciones a corta distancia. Tra ns fe re ncia de e ne rgía Convención de signos: Para sistemas cerrados, el intercambio de energía (U) sistema- +q -q entorno sólo puede ocurrir en dos formas: Calor (q) Energía que fluye a través de la frontera de un sistema en respuesta a un gradiente de temperatura. +w SISTEMA Trabajo (w) Energía que fluye a través de la frontera de un -w sistema en respuesta a una fuerza que se deplaza cierta distancia (p. ej. cuando un sistema cambia de volumen).
- 10. Primera Ley de la Termodinámica ∆U = ∆q + ∆w wrev = - P dV a P=cte En la definición de trabajo (w) se incluye un signo negativo para obtener trabajo positivo realizado sobre el sistema Para un cambio infinitesimal: dU = dq + dw dU = dq – PdV (P = cte) Si se suministra calor a un sistema (+dq), éste se expandirá (+dV), realizando trabajo sobre el entorno (- PdV). El aumento en la energía interna (U) debida al calor absorbido, es compensado por el trabajo (w) liberado hacia el entorno. Expansión isotérmica de una burbuja de gas en ascenso. Al bajar P, la burbuja se expande liberando calor al entorno (el cambio en el volumen es negativo). Para mantener T constante, el gas debe absorber calor del entorno: ∆q = −∆w ; ∆w < 0 (libera trabajo) ; ∆q > 0 (absorbe calor). ¿Qué pasaría si se aumenta la presión del sistema?
- 11. Entalpía dU = dq – PdV (P = cte) dqP = dU + PdV (dq)P : Transferencia de calor a presión constante es una variable de estado. A presión constante, w es constante y por lo tanto q debe ser también constante (1a. Ley). Se define como Entalpía (H) H = U + PV dHP = (dq)P a P constante!! dHP = dU + PdV ∆HP es una medida de la cantidad de calor CaCO3 CaO + CO2 que pierde o gana un sistema a presión ∆H constante. P . Vinicial CaCO3 CaO + CO2 Uinicial ∆H P . Vfinal P P ∆U ∆V CaO Ufinal CaCO3 + CO2 Hinicial Hfinal ∆HP = ∆U + P∆V (P = cte.) ∆HP = H productos – H reactivos
- 12. Entalpía Los siguientes procesos pueden involucrar pérdida o ganacia de calor: - Reacciones químicas como la vista anteriormente. - Un cambio de estado, p.ej. la fusión de cristales. - Un cambio de temperatura en el sistema, sin que ocurra cambio de estado, p. ej. el calentamiento de cristales. Si se suministra calor a un cuerpo de roca, la temperatura aumentará proporcionalmente de Diópsida (CaMgSi2O6) acuerdo con: ∆q = CP∆T 600 P constante Donde la constante de proporcionalidad CP es la capacidad calorífica molar a presión constante, la cual es característica del material. ∆H fusión = 144 kJ/mol El gráfico muestra los cambios en la entropía 400 H (kJ/mol) para diópsida (CaMgSi2O6) al ser calentada a P constante. Los cristales absorben calor hasta C antes del punto de fusión, y su T aumenta Cristales olº /m Líquido proporcionalmente a Cp, que define la pendiente 200 8 kJ .2 de la línea:CP= (dH / dT)P. =0 Temperatura T)P de fusión A la T de fusión, aunque se siga suministrando /d dH calor, no aumenta T, porque el calor es =( CP consumido en la ruptura de enlaces (Calor 0 latente de fusión o entalpía de fusión). Si se 0 400 800 1200 1600 sigue suministrando calor al sistema depués de T (º C) que haya fundido todo, T aumentará de manera
- 13. Enta lpía e s tá nda r de forma ción ∆Hºf Se determina experimentalmente en un calorímetro para cada sustancia. Generalmente se reporta a condiciones estándar de 298.16 K (25ºC) y 1 bar. El signo “º” indica que se trata de una sustancia pura. CaSO4 (s) + 2H2O (l) CaSO4 . 2H2O (s) anhidrita agua yeso ∆Hºf (kJ) Ca (s) + S (s) + 2O2 (g) CaSO4 (s) -1434.11 H2 (g) + 1/2 O2 (g) H2O (l) -285.830 Ca (s) + S (s) + 3O2 (g) + 2H2 (g) CaSO4 . 2H2O (s) -2022.63 Las ∆Hºf se usan para calcular la Entalpía estándar de reacción (∆Hºr) ∆Hºr = Σ∆Hºf productos – Σ∆Hºf reactivos ∆Hºr =∆Hºfyeso – (∆Hºfanhidrita + 2 ∆Hºfagua) = -2022.63 +1434.11 +(2*285.830)) Se liberan 16.86 kJ de calor por cada = -16.86 kJ mol de anhidrita que reacciona Signo – Reacción exotérmica. Se libera calor durante la reacción. Signo + Reacción endotérmica. Se debe suministrar calor para que ocurra la reacción.
- 14. S e gunda Le y de la Te rmodiná mica En un s is te ma a is la do, ca mbios e s pontá ne os ocurre n e n la dire cción de a ume nto de 2a Ley e ntropía El universo tiende a una máxima entropía (S) Esta ley introduce la función de estado S, llamada entropía, que es una medida de la uniformidad en la concentración de energía. Los procesos naturales espon-táneos tienden a igualar los gradientes de energía, o visto de otra manera, a aumentar el desorden interno del sistema. A mayor uniformidad en la concentración de la energía (o desorden del sistema), mayor será la entropía. Si se tiene un sistema aislado, con un gas en cada uno de los diferentes compartimientos. Al remover la pared que los divide, los gases se expandirán y mezclarán espontáneamente de manera irreversible. El estado final tiene mayor entropía por ser más uniforme (tener mayor desorden). Sinicial Sfinal ∆S = Sfinal - Sinicial > 0
- 15. S e gunda Le y de la Te rmodiná mica La segunda ley establece que existe una dirección natural en la que las reacciones tienden a ocurrir. Esta dirección es la de mayor entropía del sistema y su entorno. El sistema tendrá la máxima entropía en el equilibrio. dU = dq + dw dqrev = T dS a T= cte dwrev = - P dV a P= cte dU = TdS - PdV Ecuación Fundamental Te rce ra Le y de la Te rmodiná mica . La e ntropía de toda s la s s us ta ncia s pura s , cris ta lina s , pe rfe cta me nte orde na da s e s ce ro e n e l ce ro a bs oluto (0 ke lvin = -273.15ºC). La e ntropía de la s de má s s us ta ncia s e s pos itiva . Al disminuir la temperatura (o al aumentar la presión) los cristales se vuelven cada vez más ordenados, las sustituciones atómicas son menores y la entropía disminuye. (Sgas)baja P > (Sgas)alta P > Slíquido > Ssust. amorfa > Ssólido
- 16. Entropía e s tá nda r de re a cción ∆Sºr El enunciado de la 3a Ley permite determinar calorimétricamente valores absolutos de la Entropía molar estándar Sº para sustancias cristalinas puras, y a partir de los valores de Sº podemos calcular la Entropía estándar de reacción ∆Sºr . CaSO4 (s) + 2H2O (l) CaSO4 . 2H2O (s) Sº (J mol-1 K-1) anhidrita agua yeso CaSO4 (s) 106.7 H2O (l) 194.1 ∆Sºreacción = ΣSºproductos – ΣSºreactivos CaSO4 . 2H2O (s) 69.91 ∆Sºr = Sºyeso – (Sºanhidrita +2 Sºagua) = 194.1 – 106.7 – 2(69.91) = -52.42 J K-1 En termodinámica las unidades de temperatura son siempre Kelvin (K)
- 17. Ene rgía libre de Gibbs G La energía libre de Gibbs se define como: G = H – TS y dG = dH – TdS G es una variable de estado que siempre disminuye en un proceso espontáneo y alcanza el mínimo en sistemas en equilibrio. Es útil para definir la espontaneidad de una reacción, o cual lado de la reacción es más estable a ciertas condiciones de P y T. Equivale a la “energía química” de la figura. Proceso espontáneo, a P y T cte.: Si G productos < G reactivos dGT,P = G productos - G reactivos dGT,P < 0 Ghielo > Gagua Gagua > Ghielo En el equilibrio, a P y T cte.: G reactivos = G productos dGT,P = 0
- 18. G = H – TS Sustituyendo H = U + PV : G = U + PV - TS El diferencial de G es: dG = dU + PdV + VdP – TdS – SdT Sustituyendo dU = TdS – PdV dG = TdS – PdV + PdV + VdP – TdS – SdT dG = VdP – SdT Al igual que la Energía Interna (U), la Energía Libre de Gibbs (G) es una variable de estado y alcanza el valor mínimo en sistemas en equilibrio. La diferencia radica en que U alcanza el mínimo en sistemas con ciertos valores de V y S, mientras que G alcanza el mínimo en sistemas con ciertos valores de P y T. dU = TdS - PdV δG δG δU δU ----- = V ----- = - P ----- = – S ----- = T δT P δP δS V δV S T Para la reacción: CaSO4 (s) + 2H2O (l) CaSO4 . 2H2O (s) ∆Gºr = ∆Hºr – T∆Sºr ∆Hºr = -16.86 kJ = -16860 J = – 16860 –298.15(-52.42) T = 25ºC = 298.15K ∆Gºr = – 1231 J ∆Sºr = - 52.42 J K-1 ∆Gºr es negativo, por lo tanto la reacción ocurre espontáneamente. Yeso es más estable que anhidrita en presencia de agua a las condiciones de la superficie de la tierra (25ºC y 1 bar). Nota: Sólo la combinación de anhidrita + agua es metaestable, anhidrita sola no lo es.
- 19. Energía Libre de Gibbs de Formación ∆Gºf Se puede calcular la ∆Gºf de cualquier sustancia a partir de los valores de ∆Hºf y ∆Sºf . CaSO4 (s) + 2H2O (l) CaSO4 . 2H2O (s) anhidrita agua yeso Cálculo de ∆Gºf para anhidrita: Ca (s) + S (s) + 2O2 (g) CaSO4 (s) Se han calculado los Sº (J mol-1 K-1) 41.42 31.80 205.138 106.7 valores de ∆Gºf para ∆Sºf = ΣSº productos – ΣSºreactivos un gran número de sustancias y se ∆SºfCaSO4 = 106.7 – 41.42 – 31.80 – 2*205.138 encuentran en tablas. = – 376.796 J mol-1 K-1 Con estos valores se puede calcular ∆G = ∆H – T∆S directamente el cambio ∆GºfCaSO4 = ∆Hº fCaSO4 – T∆Sº fCaSO4 en la Energía Libre de = -1434110 – 298.15(-376.796) Gibbs de reacción: ∆GºfCaSO4 = -1321768 J En tablas: ∆Gºr = Σ∆Gºf productos – Σ∆Gºf reactivos ∆GºfCaSO4 = -1321790 J ∆GºfH2O = -237129 J = – 1797280 – (– 1321790) – 2(– 237129) ∆GºfCaSO4 . 2H2O = -1797280 J ∆Gºr = – 1232 J
- 20. Ene rgía libre de Gibbs pa rcia l mola r o P ote ncia l químico El potencial químico permite deducir la tendencia de flujo o reacción de material. Cuando se tienen diferencias en el potencial químico habrá flujo o reacción de material en la dirección de menor potencial. Transformación polimórfica CaCO3(s) CaCO3(s) Componentes entre calcita y aragonita Calcita Aragonita Fases Aragonita Calcita ∆µ Calcita Aragonita µ CaCO3 Calcita Aragonita Si µ c a lc it a C aC O 3 > µ a r a g o n it a C aC O 3 Material fluye de calcita a aragonita Si µ c a lc it a C aC O 3 < µ a r a g o n ita C aC O 3 Material fluye de aragonita a calcita Si µ c a lc it a C aC O 3 = µ a r a g o n it a C aC O 3 No hay flujo de material. Fases en equilibrio.
- 21. Gtot = Σ µ i Xi Xi : Fracción molar del componente i Diferencial parcial δG A Xj : Fracción molar del resto de los componentes µA = δX A P, T, X j El potencial químico de A (µ A) representa el cambio infinitesimal en G que acompaña la adición de una cantidad infinitesimal del componente A a valores constantes de P, T, y fracción molar del resto de los componentes (Xj ). El cambio total de la Energía Libre de Gibbs para el sistema petrológico más general (sistema abierto con cambio en la composición) es: Σ µi dXi = µA dXA + µBdXB + µCdXC +........ dG = VdP – SdT + Σ µ i dXi Esta ecuación muestra que G depende de: 1) Potencial químico al cambiar la concentración de los componentes (XA, XB, etc.) 2) Volumen molar, al cambiar P 3) Entropía molar, al cambiar T
- 22. De esta ecuación más general podemos sacar las siguientes conclusiones: El estado termodinámico de cualquier sistema homogéneo en el equilibrio está determinado por las propiedades intensivas: presión, temperatura y composición. Por lo tanto, la estabilidad de sistemas geológicos con variaciones en la composición depende únicamente de esas tres propiedades. El estado más estable es el de menor potencial químico µ. Si hay diferencias en los potenciales químicos de fases adyacentes, el componente de la fase con mayor potencial se moverá a la fase con menor potencial. Por ejemplo, si en un magma µ líq u id o agua > µ gas agua parte del agua se exsolverá del magma y pasará a la fase gaseosa En el equilibrio, los potenciales químicos de un componente en las diferentes fases deben ser iguales. µ líq u id o agua = µ gas agua = µ b io tita agua µ ol M gO = µ cpx M gO = µ líq . M gO
- 23. 1 componente: SiO2 APLICACIONES Cons trucción de dia gra ma s de fa s e s Los conceptos vistos se pueden representar gráficamente en un diagrama de fases. Un diagrama de fases muestra cuales fases son estables en función de la temperatura, presión, composición u otras variables. 2 componentes (sistema binario) 3 componentes (sistema ternario) CaAl2Si2O8 - CaMgSi2O6 CaMgSi2O6 - Mg2SiO4 - Mg2Si2O6 P = cte Sistema anhidro, P = cte = 20 kbar
- 24. A partir del comportamiento de la Energía Libre de Gibbs (dG = VdP – SdT) se puede predecir: a P = cte; dP = 0 a T = cte; dT = 0 δG δG dG = – SdT ------ = – S dG = VdP ------ = V δT δP Un aumento en T (+dT) hace que dG se Un aumento en P (+dP) estabiliza el vuelva más negativo para líquidos con estado de menor volumen (cristales) para entropía más alta que para cristales con minimizar G. entropía más baja. La disminución en P (-dP) estabiliza el La disminución en T (-dT) desplaza el estado de mayor volumen (líquido) para sistema al campo de estabilidad de minimizar G. cristales con menor entropía. GºL Líq GºL m = VC uid Cristales G o G m = -SL do Crista les Líqui m = VL GºC m = -SC GºC Cristales Líquido Líquido Cristales Teq. Peq. T P
- 25. Diagramas P-T e Cu sión rv ad fu o id s qu ale L Lí < G C t ris < G C C GL G GC GL = P T Superficies de Energía Libre en espacio tridimensional G-P-T. Las superficies de energía libre se intersectan en una línea curva en la que coexisten cristales y líquido en el equilibrio. La proyección de esta línea en forma paralela al eje G sobre el plano P-T (flechas) es la curva de fusión mostrada a la derecha. A baja T, la superficie de energía libre del líquido se localiza a mayor G que la del equilibrio (cristales son más estables). A alta T, la superficie de energía libre del líquido se localiza a menor G que la del equilibrio (líquido es más estable). En un diagrama P-T, para cualquier valor de presión y temperatura se tiene la mínima Energía Libre de Gibbs (G), i.e., se representan las fases estables.
- 26. P rincipio de Le Cha te lie r: e Si un cambio ocurre en un sistema, el sistema Curva d fusión responde de manera que la fuerza que causa el cambio sea minimizada o moderada. Cristales GC < GL P Si aumenta P, el sistema responde disminuyendo V. +P Líquido A alta P serán más estables las fases que GL < GC ocupen menor volumen molar (cristales). -T +T Al bajar P, el líquido, que generalmente ocupa mayor volumen molar, será más -P estable (ej. fusión por decompresión). Si aumenta T el sistema responde T aumentando S. A alta T serán más estables los estados más En la curva de fusión coexisten desordenados (líquido tiene mayor entropía). cristales y líquido, A baja T serán más estables los estados más GL = GC (∆G = 0) ordenados (cristales tienen menor entropía) SL > SC (∆S > 0) VL > VC (∆V > 0) La curva en este caso debe tener pendiente positiva (al aumentar P, la temperatura de fusión se desplaza a valores más altos). Este Los diagramas de fases también se caso es común en minerales y rocas. determinan experimentalmente.
- 27. P e ndie nte de la curva En el equilibrio: GC = GL Al variar P y T, pero permaneciendo en La ecuación de Clausius-Clapeyron la curva de equilibrio, el cambio en G de permite calcular la pendiente de la curva las dos fases debe ser igual: de equilibrio en un diagrama P-T a partir de valores de volumen molar y entropía dGC = dGL a diferentes valores de P y T. A B VCdP – SCdT = VLdP – SLdT V ∆ S/∆ (VC – VL )dP = (SC – SL)dT m= dP (SC – SL) B ---- = ----------- dT (VC – VL ) A dP dS ---- = ---- dT dV T Ecuación de P Clausius-Clapeyron
- 28. De pe nde ncia de T y P En tablas se reportan valores para H, S, G, V a condiciones estándar (25 ºC y 1 bar), pero en problemas petrológicos nos interesa conocer las condiciones de equilibrio a otros valores de P y T, para establecer la posición de una curva de reacción en un diagrama P-T (no sólo su pendiente). (No hay apuntes de esta sección)
- 29. Ejercicio 4: Transformación polimórfica de CaCO3 CaCO3 CaCO3 Calcita Aragonita ∆ H ºf ∆G ºf Sº Vº Componente Fase kJmol-1 Jmol-1K-1 cm3mol-1 CaCO3 Calcita -1206.9 -1128.8 92.9 36.934 CaCO3 Aragonita -1207.1 -1127.8 88.7 34.150 Parámetros reportados a condiciones estándar: 298.16 K (25ºC) y 1 bar. El signo “º” indica que se trata de una sustancia pura. Calcular ∆Hºr ∆Gºr ∆Sºr ∆Vºr ¿Cuál de las dos fases será estable a condiciones atmosféricas? Efecto de P y T (pendiente de la curva en diagrama P-T)
- 30. Ejercicio 4: Transformación polimórfica de CaCO3 CaCO3 CaCO3 Calcita Aragonita A 1 bar y 25 ºC ∆GºfCc < ∆GºfA ∆Gºr (prod. - react.) es positiva GA = GCc Reacción no ocurre espontá- SºCc > SºA neamente; Calcita es más VºCc > VºA estable a 1 bar y 25ºC SºCc > SºA ∆Sºr es negativo Aragonita dG = -S dT GA < GCc m = dP/dT = ∆S/∆ V Un aumento en T favorece a P Calcita (con mayor S) (dG se vuelve más negativo) Calcita VºCc > VºA ∆Vºr es negativo GCc < GA dG = VdP Un aumento en P favorece a T Aragonita (con menor V) (dG se vuelve más negativo) PENDIENTE POSITIVA !!
- 31. Eje rcicio 5 CaCO3 CaCO3 Calcita Aragonita ∆ H ºf ∆G ºf Sº Vº Componente Fase kJmol-1 Jmol-1K-1 cm3mol-1 CaCO3 Calcita -1206.9 -1128.8 92.9 36.934 CaCO3 Aragonita -1207.1 -1127.8 88.7 34.150 6000 5000 4000 P (bar) 3000 Aragonita GA < GCc 2000 1000 Calcita GCc < GA 0 0 100 200 300 400 500 T (K)
- 32. Eje rcicio 6: Tra ns forma ción P olimórfica de Al2S iO 5 Dis te na (Cia nita )-Anda lus ita -S ilima nita Datos termodinámicos Fase ∆ H ºf ∆ G ºf Sº Vº kJ/mol kJ / mol J/mol K cm3 / mol Distena -2594.29 -2443.88 83.81 44.09 Andalusita -2590.27 -2442.66 93.22 51.53 9 Silimanita -2587.76 -2440.99 96.11 49.9 8 7 6 Distena P (Kb) 5 4 3 Silimanita 2 1 0 Andalusita 100 200 300 400 500 600 700 T (ºC)