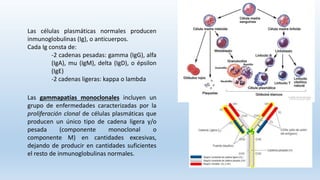
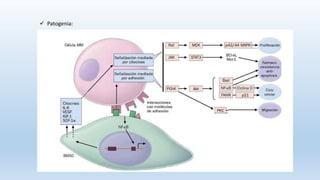
Las gammapatías monoclonales incluyen un grupo de enfermedades caracterizadas por la proliferación clonal de células plasmáticas que producen un único tipo de cadena ligera y/o pesada. El mieloma múltiple constituye una proliferación maligna de células plasmáticas derivadas de un solo clon que puede ocasionar dolores óseos, anemia, insuficiencia renal, infecciones y otras complicaciones. El diagnóstico requiere la detección de un componente monoclonal en sangre o médula ósea así como lesiones osteol