Descargado 116 veces

























































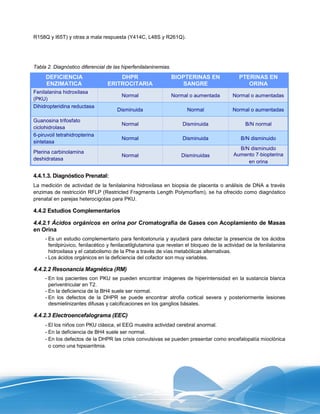
















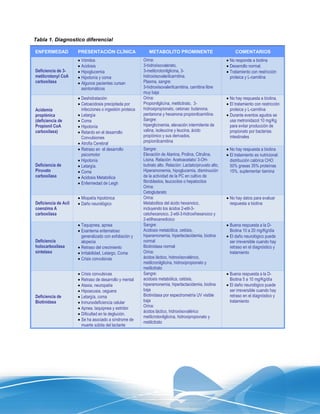










Este documento presenta las líneas generales para la detección, seguimiento y control de cuatro enfermedades metabólicas congénitas (hipotiroidismo congénito, hiperplasia adrenal congénita, fenilcetonuria y deficiencia de biotinidasa) en el Instituto Mexicano del Seguro Social. Describe los objetivos, metas y estrategias para realizar la detección de estas enfermedades mediante pruebas en muestras de sangre de talón de recién nacidos, así como el seguimiento y tratamiento oport



























































