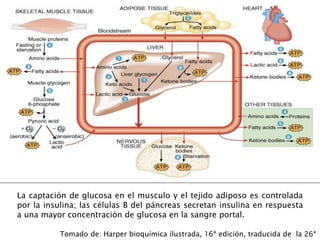
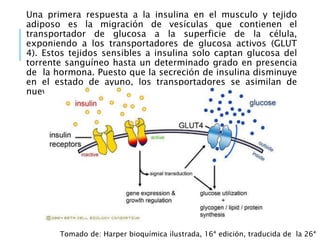
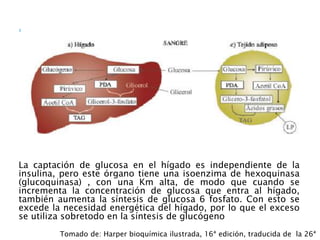
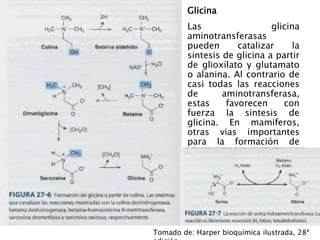
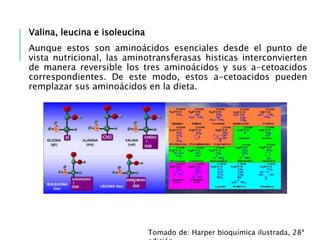
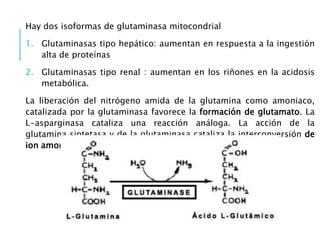
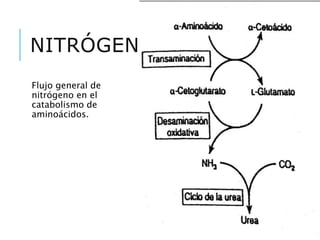
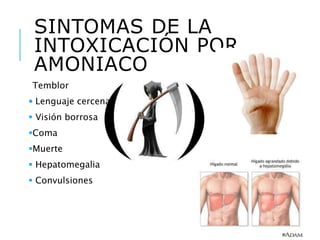
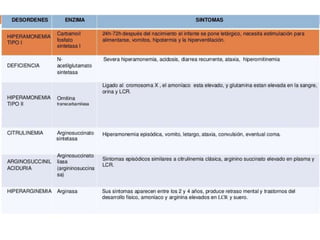
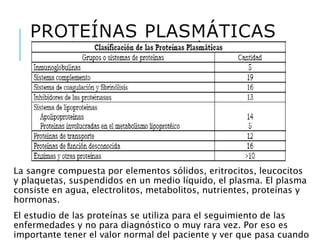
El documento resume los principales procesos metabólicos relacionados con los carbohidratos, lípidos y proteínas en el cuerpo humano. Explica cómo estos nutrientes se integran y convierten entre sí para proporcionar energía a los tejidos y ser almacenados o eliminados, dependiendo de si la persona se encuentra alimentada o en ayunas. También describe cómo las hormonas como la insulina y el glucagón controlan estos procesos y cómo trastornos como la diabetes afectan el metabolismo.