Descargado 26 veces




































![SesióndeBioinformática.Basesdedatosyherramientasbioinformáticas|práctica5
37
841 tcttttataa taaactaatg ataactaatg acatccagtg tctccaaaat tgtttccttg
901 tactgatata aacacttcca aataaaaata tgtaaatgag tggttaatct ttaaaaaaaa
961 aaaaa
//
Con la secuencia del gen, ya sea en forma de secuencia completa con intrones o en la forma de
rna mensajero, se iniciará una búsqueda de secuencias relacionadas en otros genomas, con la
finalidad de explorar el grado de divergencia de ambas secuencias o de comparar la presencia
de inserciones o deleciones de nucleótidos a lo largo de la evolución de ambas secuencias a
partir de un ancestro común. Para esto el Ncbi cuenta con un programa de comparación de
secuencias denominado blast (Basic Local Alignment Search Tool o Herramienta Básica de Bús-
queda por Alineamiento Local). Este algoritmo está basado en la comparación de similitudes
de nucleótidos o aminoácidos entre secciones cortas (locales) de la secuencia de búsqueda
(query) y cada una de las secuencias de la base de datos; para proteínas, la longitud de las se-
cuencias comparadas es de tres aminoácidos y para nucleótidos la longitud mínima es de 11.
Cuando el algoritmo encuentra regiones idénticas o similares, amplía la búsqueda en regiones
contiguas, extendiendo el alineamiento hasta que ya no encuentre identidades o similitudes y
evaluando el grado de sustituciones o de inserciones y deleciones de nucleótidos o aminoáci-
dos entre las secuencias. Al seleccionar en la página de entrada la opción blast, aparecerá una
ventana con la siguiente información:
En esta página se selecciona nucleotide blast para realizar la búsqueda con una secuencia de
nucleótidos.
Aparecerá una ventana en la que se inserta la secuencia del mRna obtenida anterior-
mente. Más abajo, en selección de programa, conviene seleccionar blastn (somewhat similar
sequences [blast n]) para permitir una búsqueda amplia.
Entre los parámetros opcionales más importantes del algoritmo figuran:
Expect threshold: Número de secuencias que pueden ser encontradas al azar (Valor de
Expect con elevada significación estadística: 0.000001 = (1 x 10-6
)).
Word Size: La longitud de la secuencia que comienza el alineamiento entre secuencias
relacionadas.](https://image.slidesharecdn.com/biotecnologiav14-141116232701-conversion-gate01/85/Biotecnologia-v14-37-320.jpg)

























![Degradacióndecompuestosxenobióticosatravésdeactividadesenzimáticas|práctica10
63
4. Se vacían ~ 10 ml de medio con petróleo en cada caja Petri (45 mm de diámetro) y lo
mismo del medio sin petróleo.
5. Los micelios de los dos tipos de hongos se siembran tomando un fragmento de 4×4
mm de la caja original, inoculando por triplicado en las cajas V8 con y sin petróleo
bajo condiciones de esterilidad.
6. Los hongos se dejan crecer 30 días a temperatura ambiente y se observa el resultado.
Nota: Para llevar a cabo la segunda parte se utilizarán los hongos tolerantes y no tolerantes
cultivados en medio V8 de esta primera parte de la práctica.
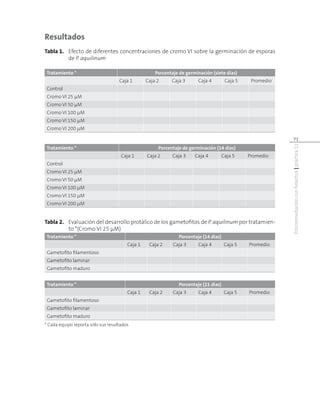
Resultados
La cepa del hongo tolerante a petróleo deberá crecer de manera similar a la condición control,
mientras que el crecimiento de la cepa no tolerante se notará afectado.
Anotar las observaciones, teniendo en cuenta los siguientes puntos:
1. ¿Qué mecanismos son los responsables de comportamientos diferentes en los hon-
gos bajo estas condiciones?
2. ¿Qué efectos tendrá el petróleo en los organismos vivos?
Parte II. Actividad enzimática
Los hongos cuentan con diversas enzimas extracelulares que les permiten degradar ciertos
compuestos, entre estas enzimas se encuentran las lacasas, que están involucradas en la
transformación de una amplia variedad de compuestos fenólicos.
Con base en esto se pretende probar la capacidad ligninolítica de estos hongos a través de
un ensayo colorimétrico, en donde un sustrato es oxidado (cambiará el color del sustrato como
indicador) en presencia de la enzima lacasa.
Material y equipo
1. Preparación del medio mineral 20X + ABTS [1mM] modificado de Olsson, 2001.
Composición del medio mineral 20X (stock 1 L):
Reactivo Por litro de volumen final
K2
HPO4
2 g
MgSO4
·7H2
O 2 g
KCl 2 g
En un vaso de precipitados, preparar:
Reactivo Por litro de volumen final
Stock medio
mineral 20X
50 ml
ABTS [1mM] 0.5487 g
Agar 20 g](https://image.slidesharecdn.com/biotecnologiav14-141116232701-conversion-gate01/85/Biotecnologia-v14-63-320.jpg)
![Manualdebiotecnología|práctica10
64
Nota: Este medio es sólo para la determinación de actividad, la abundancia del hongo no es
relevante.
2. Aforar a 1 litro con agua destilada. Transferir a un matraz Erlenmeyer y esterilizar
durante 15–20 min. a una temperatura de 120 o
C y 1 atm de presión. Vaciar 10 ml de
medio en cada caja Petri (45 mm de diámetro). Colocar a 4 o
C en oscuridad (envolver
con papel aluminio).
3. Se toman micelios de los hongos ya crecidos en las cajas con medio V8 y se cortan
en cuadros de 1 cm2
; se inoculan en el medio mineral 20X + ABTS [1mM]. Se incuban
durante una semana a 28 o
C. en ausencia de luz, cubriendo las cajas con papel alu-
minio. El ensayo se lleva a cabo por triplicado.
Resultados
El ABTS oxidado por la lacasa se vuelve visible cuando el medio cambia la coloración a verde o
azul oscuro, por lo que la cepa del hongo tolerante a petróleo deberá hidrolizar al ABTS, mien-
tras que la cepa no tolerante no tendrá reacción alguna.
De acuerdo con lo observado, responder:
1. ¿Qué relación tiene el ABTS con el petróleo?
2. ¿Por qué cambia de color el ABTS en presencia de la lacasa?
Bibliografía
Baldrian P. (2006), “Fungal laccases - occurrence and properties”, FEMS Microbiol 30: 215-242.
Field, J.A., E. De Jong, G. Feijoo-Costa, y J.A.M. De Bont (1993), “Screening for ligninolytic fungi
applicable to the biodegradation of xenobiotics”. Trends Biotechnol 11: 44-49.
Rojas-Avelizapa, N., E. Olvera-Barrera y L. Fernández-Linares (2005), “Feasibility study of biore-
mediation of a drilling-waste-polluted soil: Stimulation of microbial activities and hydro-
carbon removal”. Journal of environmental science and health, Part A, Toxic/hazardous
substances & environmental engineering 40: 2189-2201.
Margesin, R., A. Zimmerbauer y F. Schinner (2000), “Monitoring of bioremediation by soil biolo-
gical activities”, Chemosphere 40: 339-346.](https://image.slidesharecdn.com/biotecnologiav14-141116232701-conversion-gate01/85/Biotecnologia-v14-64-320.jpg)

































Este documento presenta el protocolo de una práctica de laboratorio sobre la fermentación alcohólica mediada por Saccharomyces cerevisiae. La práctica consta de cuatro partes: 1) preparación del medio de cultivo a partir de uvas, 2) inoculación e incubación para permitir la fermentación, 3) obtención del producto final mediante filtración y centrifugación, y 4) análisis bioquímico para determinar la concentración de azúcares y alcohol producido. El objetivo es familiarizar a los estudiantes con




![Ppt biokimia i_(biomembran)[1]](https://cdn.slidesharecdn.com/ss_thumbnails/pptbiokimiaibiomembran1-160417123031-thumbnail.jpg?width=640&height=640&fit=bounds)










































