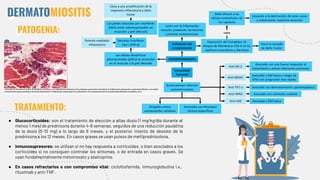
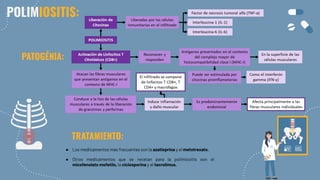
El documento describe las miopatías inflamatorias idiopáticas, centrándose en dermatomiositis y polimiositis, que se manifiestan por debilidad muscular y pueden tener implicaciones sistémicas. Se detallan aspectos epidemiológicos, patológicos y diagnósticos, indicando un tratamiento basado en glucocorticoides y otros inmunosupresores. El diagnóstico se apoya en la presencia de autoanticuerpos específicos y resultados de biopsia muscular.