




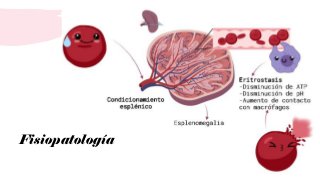


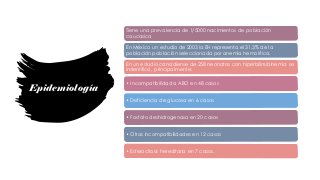
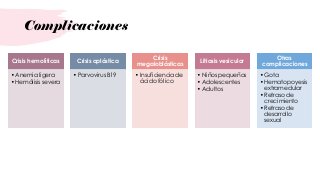



La esferocitosis hereditaria es una enfermedad hereditaria causada por deficiencias moleculares en proteínas de la membrana eritrocitaria como la espectrina, ankirina y banda 3. Esto causa fragilidad osmótica de los eritrocitos y anemia hemolítica. Se hereda de forma autosómica dominante y su prevalencia es de 1 en 2000 personas. Los síntomas van desde asintomáticos hasta anemia severa, requiriendo tratamiento con ácido fólico y esplenectomía en casos graves.



































































